




Myelopathy is a common diagnosis often made by clinical neurologists and can be a cause of significant disability. Differential diagnosis classically includes structural etiology, demyelinating, metabolic, nutritional, inherited, and degenerative causes, resulting in a vast array of pathophysiologic processes, underlying myelopathy (Table 1). It is critical that clinicians use their clinical knowledge and utilize appropriate diagnostic studies in order to arrive at the correct diagnosis. This will ensure appropriate and timely management and counseling. Here, we present and discuss three unusual cases presenting with myelopathy, within a three-month period, and their respective workup and uncommon diagnoses. All patients were seen at the Neuromuscular Division of the Department of Neurology at the University of Southern California Keck Medical Center, US. Each of these cases reveals an unexpected diagnosis and highlights the importance of a broad differential diagnosis when confronted with a patient who may exhibit signs of myelopathy.
Case Presentations
Case One
History
A 44-year-old female physician with a history of hyperthyroidism was referred to our neuromuscular division for evaluation of abnormal gait, right leg weakness and cramping of five weeks’ duration. The patient had recently vacationed in Africa, when she noted the occurrence of pain in her right thigh, followed by a noticeable limp in her gait. Over the next few weeks, she noted worsening in her gait, the inability to run or walk long distances, and the occurrence of cramping pain in her legs. She denied back or neck pain, radicular pain, bowel or bladder symptoms, cranial nerve related complaints, or other neurologic deficit. As a physician, she became extremely concerned and anxious that she might be developing a serious, progressive, degenerative neurological ailment such as multiple sclerosis or amyotrophic lateral sclerosis.
Physical Examination
Pertinent findings on examination revealed increased muscle tone, predominantly in the right lower extremity, mild right hip flexion, knee flexion, and ankle dorsiflexion weakness. There was slight decrease in joint position and vibration sensation at the toes bilaterally. She exhibited a spastic gait on the right with inability to heel walk or tandem walk. She had difficulty squatting. She was diffusely hyper-reflexic with sustained right ankle clonus, positive crossed adductor, suprapatellar, and pectoralis reflexes, bilateral plantar extensor responses, and positive Hoffmann’s signs.
Diagnostic Studies
Initial workup including thyroid-stimulating hormone (TSH) blood test, vitamin B12 level, methylmalonic acid, homocysteine, serum copper, and human T-lymphotropic virus 1 (HTLV-1) antibody were all normal. Magnetic resonance imaging (MRI) of the brain, cervical and thoracic spine, with and without contrast, was performed.
Imaging
A previous MRI, which was done at an outside facility, of her lumbar spine revealed moderate canal stenosis at L2-S1 with some ossification of the
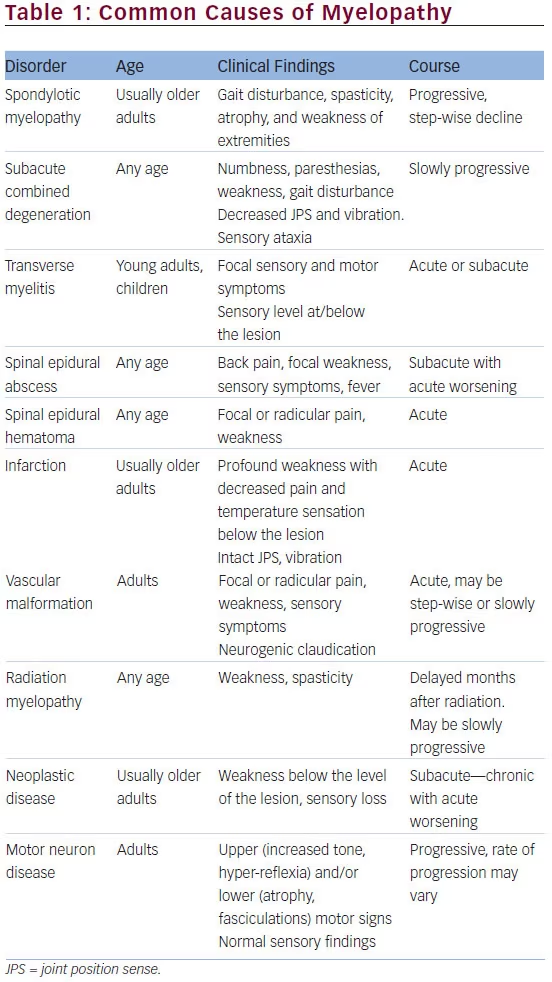
posterior longitudinal ligament. MRI of her cervical/thoracic spine revealed significant ossification of the posterior longitudinal complex enlargement resulting in severe central canal stenosis and multilevel cord compression from C2-3 to T2-3. Images are shown below (Figures 1 and 2).
Diagnosis
Cervical myelopathy from ossification of posterior longitudinal ligament (OPLL).
Clinical Course
The patient underwent a C2 to T4 laminectomy and posterior spinal fusion. She tolerated the procedure without any complication and is recovering well. Her right leg strength has dramatically improved to the point where she is able to walk without any assistive device and is able to ambulate independently with minimal difficulty. On the most recent examination she has hyper-reflexia but no longer sustained
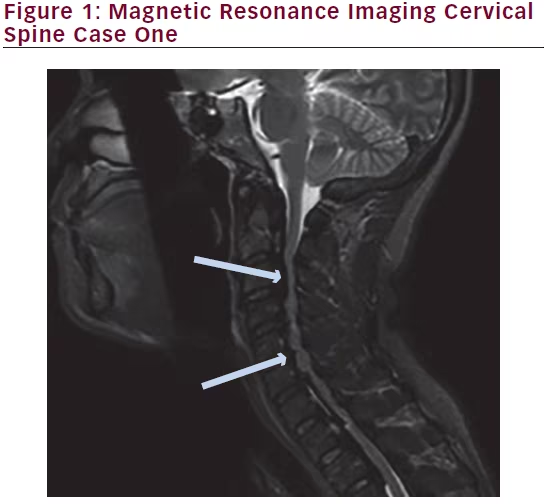
clonus of the right lower extremity. She has recovered well enough to return to work.
Case 2
History
A 28-year-old male without prior medical history presented for evaluation of chronic, progressive gait disturbance. Four years prior, the patient noticed stiffness in his legs and difficulty running. Over the following years he developed clumsiness of his legs, such that he would trip on carpet or bump into objects while walking. Two years prior, he lost the ability to run. Subjectively, he complained of decreased sense of balance
while standing. He had been started on baclofen for spasticity but this was only mildly helpful. There was no notable family history for gait disturbance or other neurologic deficits.
Physical Examination
Pertinent findings on examination revealed sparse hair, normal skin color for race, normal motor strength, increased muscle tone in the lower extremities, and a slightly decreased vibratory sensation at the toes. The remainder of his sensory examination was normal. Deep tendon reflexes were diffusely hyperactive, with sustained bilateral ankle and patellar clonus. Extensor plantar responses and bilateral Hoffman’s signs were present. His gait was spastic, with scissoring noted in the left leg. He was unable to tandem or heel walk.
Diagnostic Studies
MRI of the thoracic spine from an outside facility reportedly showed midthoracic cord atrophy. Nerve conduction study (NCS) of the right lower extremity and the left upper extremity revealed normal sensory and motor responses. Posterior tibial H-reflexes were symmetric but of large amplitude at subthreshold stimulation. Electromyographic examination revealed no evidence of fibrillation potentials, positive sharp waves, fasciculations, or neurogenic changes. Insertional activity was also normal. Serum copper, methylmalonic acid, and homocysteine were normal. HTLV- 1 and HTLV-2 antibodies were negative. Very long chain fatty acids, namely, tetracosanoic acid (C24:0), hexacosanoic acid (C26:0), and their ratios to docosanoic acid (C22:0) of the serum were elevated. Genetic testing revealed a 44 base pair deletion in ABCD1 gene resulting in a hemizygous frameshift mutation of a member protein of the adenosine 5’-triphosphate (ATP) binding cassette family. This mutation is thought to be responsible for deficiencies in transporting very long-chain fatty acids (VLCFAs) into peroxisomes. As a result, this and other mutations in the ABCD1 gene result in fatty acid accumulation in tissue. This particular frameshift mutation is expected to be inherited in an X-linked recessive fashion.
Diagnosis
X-linked adrenomyeloneuropathy (AMN).
Clinical Course
The patient will be starting medical therapy with Lorenzo’s Oil and will be followed for signs of clinical improvement or progression. He will also be undergoing extensive physical therapy for gait training and mobility. He will be followed closely with yearly MRIs of the brain to look for signs of cerebral demyelination.
Case 3
History
A 67-year-old female with a history of diabetes, previous gastric bariatric surgery, and a subsequent weight loss of more than 150 pounds presented for evaluation of gait abnormality and sensory changes. Her bariatric surgery was performed over seven years ago, however, she had a recent partial gastrectomy to treat her gastroesophageal reflux disease (GERD) symptoms four months prior to symptom onset. She developed progressive problems with balance, which was worse in the dark, and she suffered from falls, which resulted in a fractured shoulder. Her gait deteriorated to the point where she required a wheelchair for mobility. The patient described problems with tingling in her fingers, particularly in the index fingers and thumbs of both hands, and in the lower extremities extending from the toes to the mid-thighs. Her course was initially progressive but appeared to stabilize in the months prior to her initial presentation.
Physical Examination
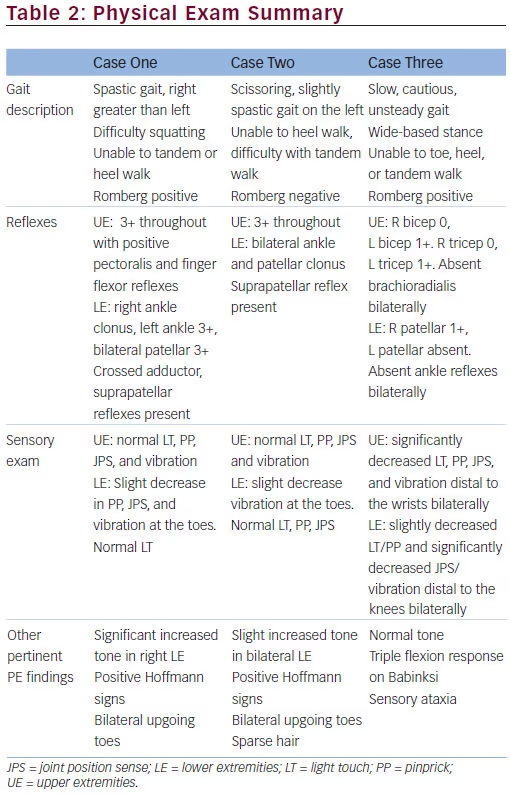
Pertinent findings on examination revealed significantly decreased vibratory sensation in all four extremities and decreased position sense in the toes bilaterally. She had mild weakness in her bilateral hip flexors, wrist extensors, knee extensors, and shoulder abductors. Deep tendon reflexes were absent at the ankles and brachioradialis bilaterally. Babinski testing revealed a triple flexion response. Her gait was cautious and unsteady. Romberg testing showed prominent truncal ataxia. She was unable to toe, heel, or tandem walk. She exhibited a wide-based stance.
Diagnostic Studies
Initial workup including vitamin B12, TSH, hemoglobin A1c, and selenium levels were normal. Preliminary serum copper and zinc levels approximately six months prior to arrival in the neurology clinic were 8 mcg/dL (lower limit of normal = 70 mcg/dL) and 157 mcg/dL (upper limit of normal = 130 mcg/dL), respectively. When repeated at time of neurologic evaluation, these values were again abnormal at 6 mcg/dL and 168 mcg/dL, respectively. Imaging studies were also pursued. Electrodiagnostic studies revealed a mild length-dependent axonal sensorimotor neuropathy. Antibodies to aquaporin 4 were negative, as imaging may have been consistent with neuromyelitis optica.
Imaging
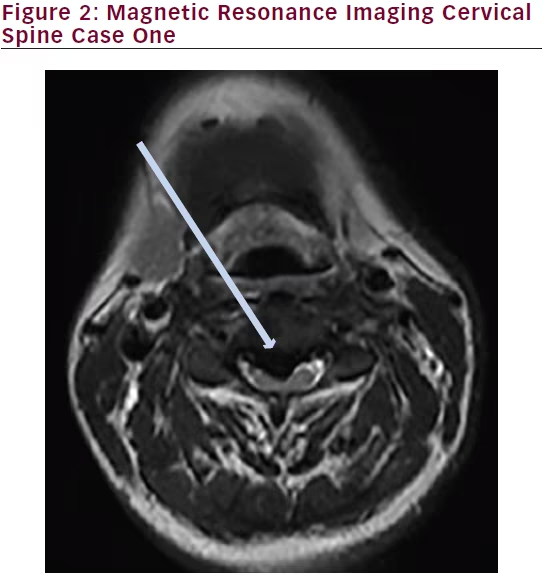
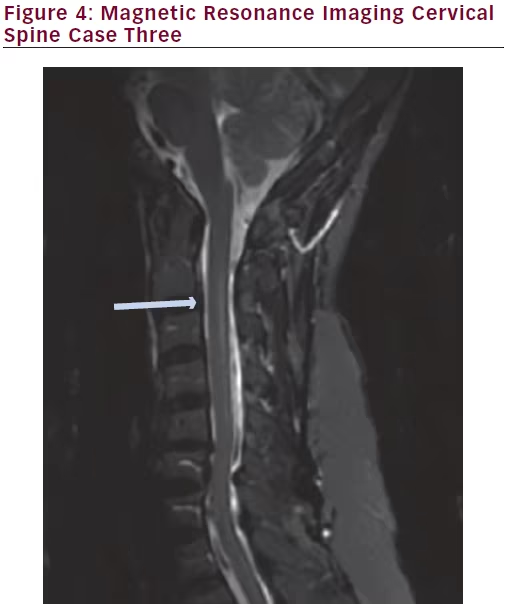
MRI of the brain with and without contrast was unremarkable. MRI of the cervical spine without contrast revealed T2 hyperintensity within the posterior central aspect of the cord from the C2 to the C6 level. Images are shown below (Figures 3 and 4).
Diagnosis
Copper deficiency myelopathy.
Clinical Course
The patient had been treated with vitamin B12 injections and elemental copper supplementation prior to our initial neuromuscular evaluation. She was also instructed to limit her zinc intake. Her clinical course, although initially progressive, had stabilized and indeed the sensory symptoms in her upper extremities have improved since the initial evaluation. She will be followed for signs of clinical improvement and serial serum copper evaluations. Physical exam findings for each case are summarized in Table 2.
Discussion
Case One—Ossification of Posterior Longitudinal Ligament
OPLL is an important cause of myelopathy that results from the ossification of the posterior longitudinal ligament (PLL), usually in the cervical region, but may extend into the thoracic and lumbar regions as well. The PLL extends along the posterior surface of the vertebral bodies adjacent to the anterior aspect of the spinal cord. In some patients, the PLL ossifies and hypertrophies, progressively shortening the spinal canal diameter. As ossification progresses, it may cause myelopathy via two different mechanisms. The first is due to compression of the hypertrophied PLL on
the anterior cord, leading to direct mechanical damage to the cord. The second is vascular damage due to mechanical vascular compression and localized ischemia. This progressive shortening of the spinal canal diameter due to PLL ossification may predispose patients to mechanical spinal cord injury even after relatively minor trauma.1,2
For the patient in Case One, the initial differential diagnosis included multiple sclerosis given the patient’s sex, age, and physical examination findings. Although she was spared from a diagnosis of a progressive, degenerative neurologic condition, the patient required an extensive C2-T4 laminectomy and posterior spinal fusion. Post-operatively she has made a strong recovery,
and is close to her previous level of mobility and overall functionality. In this case, imaging was pursued to evaluate for demyelinating disease or other structural or space-occupying lesion, making the ossification of the PLL an unexpected discovery. Identifying this rare diagnosis allowed the patient to have the appropriate and amenable surgical intervention.
Case Two—X-linked Adrenomyeloneuropathy
AMN is an X-linked recessive disorder that is present almost exclusively in males, although a minority of cases are female. Mutation in the ABCD1 gene causes deficiency in adrenoleukodystrophy (ALD) protein, which normally functions to transport VLCFAs into peroxisomes for metabolism via beta-oxidation. This deficiency causes accumulation of VLCFA in several body tissues including the nervous system, adrenal cortex, and testes.3 The accumulation in the spinal cord causes a noninflammatory distal axonopathy involving the long tracts of the spinal cord and peripheral nerves.4 When cord axon degeneration occurs, symptoms of myelopathy develop, including spasticity, weakness, sexual dysfunction, ataxia and distal polyneuropathy.
There are three common clinical patterns. The most prevalent type, Adrenomyeloneuropathy, accounts for 40–60% of cases. Its typical onset is in the third decade, with slowly progressive gait disturbance and spastic paraparesis.5,6 Secondly, the classic childhood cerebral type presents at age four to eight years with encephalopathy, cognitive and behavioral abnormalities, and clinical regression of developmental milestones.7 The least common type of AMN may present with only Addison’s disease. These presentations may be dynamic; approximately 20% of AMN males will progress to cerebral demyelination type. If progression to cerebral ALD is detected, early allogeneic hematopoietic stem cell transplantation is a therapeutic option to arrest the progression. However, once progression is complete, there are no proven effective treatments.4 There is one medication, Lorenzo’s Oil that has been proposed as a treatment for AMN. It is composed of a 4:1 mixture of oleic acid and erucic acid. Multiple studies have shown that Lorenzo’s Oil reduces the levels of VLCFA, though it has no proven efficacy in symptomatic AMN.8 There is one retrospective study that showed efficacy in preventing neurologic progression in asymptomatic boys.9 Nonetheless, treatment with Lorenzo’s Oil will be pursued and the patient will be followed for signs of clinical improvement. More importantly, the patient will be evaluated with yearly MRIs of the brain to monitor for signs of cerebral demyelination, which could mark the progression to cerebral ALD. If and when the MRI points to progression to cerebral ALD, hematopoietic stem cell transplantation may be pursued.
Case Three—Copper Deficiency Myelopathy
Copper is an important cofactor in many enzymes associated with the maintenance of myelin structure and function. These enzymes include cytochrome-c-oxidase, copper/zinc superoxide dismutase, tyrosinase, dopamine beta-hydroxylase, lysyl oxidase, peptidylglycine alphaamidating monooxygenase, monoamine oxidase and ceruloplasmin.10 These enzymes are critical to maintaining the myelin sheath. Without these enzymes, demyelination occurs most often in the dorsal columns similar to severe combined degeneration. Demyelination of the lateral columns may also occur. As spinal cord demyelination occurs, patients develop sensory ataxia, spasticity, gait disturbance, impaired vibration, and proprioception sensation. Patients may also develop signs of peripheral neuropathy, including lower extremity paresthesias, pain and temperature sensation impairment, and decreased deep tendon reflexes. The peripheral neuropathy may occur at same time or possibly prior to the myelopathy, and reflexes may either be brisk or depressed due to the simultaneous existence of both.11
The patient in Case Three presented with peripheral neuropathy with hypo-reflexia, despite imaging, which revealed cervical cord signal changes. This discrepancy highlights the potentially difficult task of lesion localization in a patient with copper deficiency, as they may be either hyper-reflexic from myelopathy or hypo-reflexic from co-existing neuropathy. Bariatric surgery is designed to impair the absorption of nutrients to prevent or reverse obesity. However, bariatric surgery patients are at particularly elevated risk of copper, vitamin B12, vitamin D, and vitamin E deficiency from impaired dietary nutritional absorption that occurs in the months to years that follow surgery.12 Often, the presentation of copper deficiency is insidious, without clear neuropathic or myelopathic findings for several months to years post-operatively. Another common cause of copper deficiency is excessive zinc, which may be a result of nutritional supplements or denture cream use. Zinc inhibits intestinal absorption of copper, and chronic high-dose zinc supplementation may result in depressed serum copper levels.13 Our patient had previous gastric bypass surgery, chronically low copper and high zinc levels. These latter laboratory findings were previously addressed with copper supplementation and a decrease in her zinc supplementation by her surgeon. Despite appropriate copper supplementation, the patient was still symptomatic with measurably low copper levels in her serum.
Conclusion
These three cases demonstrate the broad spectrum of pathology that may be responsible for the clinical signs of myelopathy. Whether it is structural, genetic, metabolic, or nutritional in nature, it is critical for the clinician to maintain and investigate a broad differential diagnosis to arrive at the correct diagnosis and provide appropriate treatment and intervention.


