Background for molecular-based therapy for spinal muscular atrophy
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disease and the most common cause of infant death worldwide, with an incidence of 1:10,000 live births and carrier frequency of 1:50.1 This disease, caused by degeneration of spinal and bulbar motor neurons, is characterized by progressive muscle weakness and atrophy, scoliosis and feeding and respiratory problems.
Unravelling the complexity of SMA was a collaboration between insightful clinicians and laboratory scientists. The clinical spectrum includes observations reaching back over 100 years.2 The first description, attributed to Werdnig in 1891, included two brothers aged 10 months.3 Hoffmann’s 1893 review added seven cases of his own.4 A coincident description by Thomson and Bruce, also in 1893, brought a disease of intermediate severity with prominent scoliosis into the clinical spectrum.5 There was then a long hiatus until 1956 when Kugelberg and Welander described the third major clinical variant of SMA, previously thought to be a form of muscular dystrophy.6 Four decades later, in 1991, the International SMA Consortium on Childhood SMA classified patients into the three clinical groups that we recognize today (Table 1).7,8
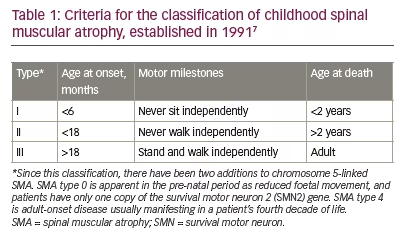
The acute form of type 1 SMA (Werdnig–Hoffmann disease) is characterized by severe generalized muscle weakness and hypotonia at birth or within the first 6 months, and is usually followed by death within 2 years. Children with type 2 SMA (Dubowitz disease) can sit, although they cannot stand or walk unaided, and survive beyond 2 years. In type 3 SMA (Kugelberg–Welander disease), patients have proximal muscle weakness, starting after the age of 18 months. In practical terms, clinical severity shows a continuous spectrum from mild to very severe SMA.9
The unknown underlying biochemical defect made identifying the gene for SMA more challenging. Shortly after the clinical phenotypes were identified, Melki et al. mapped the chromosomal linkage of the three forms of SMA to chromosome 5 (5q11.2-13.3).10,11 Further characterization of the SMA locus revealed highly homologous duplications of the SMN1 telomeric and SMN2 centromeric regions. Both genes are transcribed. Five nucleotides distinguish the paralogous SMN genes and account for the alternative splicing with loss of exon 7 specific to SMN2 transcripts (Figure 1).12,13 Full-length transcripts are almost exclusively produced by SMN1. Lefebvre et al. discovered that SMN1 was lacking in 98% of patients with SMA.12 SMN2 is unable to compensate for the SMN1 loss. Therefore, the development of the SMA phenotype is caused by two events: inherited (or de novo) SMN1 gene mutations; and a constitutive defect of the SMN2 gene leading to less full-length SMN protein being produced.12
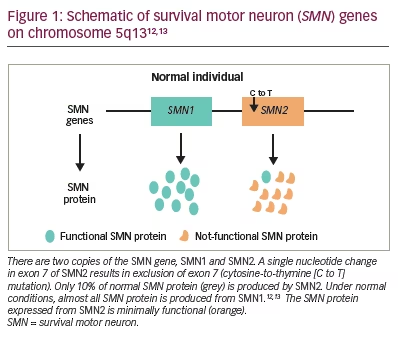
Unravelling the clinical phenotypes and genetics of SMA represent the first steps in optimizing newborn screening and the treatment of this complex disease.
Efforts to treat spinal muscular atrophy based on antisense oligonucleotide technology
Following gene discovery, the first molecular efforts to treat SMA focused on correcting the SMN2 gene to compensate for the non-functional SMN protein. A single nucleotide mutation adversely affects splicing in exon 7 of SMN2. Blocking this mutation by antisense oligonucleotide therapy restores SMN2 during pre-mRNA splicing.14 Proof-of-concept studies showed efficacy in a transgenic mouse (D7) expressing human SMN2. The SMNΔ7 neonatal mouse model is also genetically smn null and has a lifespan of ~15 days.15,16 This enabled clinical trials of the antisense oligonucleotide nusinersen.17,18
In the randomized, double-blind, sham-controlled, phase III ENDEAR trial, nusinersen was administered by intrathecal injection.17 Four loading doses were given and the first three doses administered at 14-day intervals.17 The fourth dose was administered 30 days after the third dose. Continued dosing was maintained every 4 months for the duration of the 13-month trial. The sham procedure consisted of a small needle prick to the skin over the lumbar spine. A total of 122 symptomatic infants were randomized; 81 were assigned to the nusinersen group, and 41 to the control group. All had weakness onset by 6 months.
The trial had two primary efficacy endpoints: a motor milestone response, which was defined according to results on the Hammersmith Infant Neurological Examination (HINE) (Section 2, HINE-2; scoring patient according to motor skills on eight items [Table 2]18,19 and event-free survival.) This was defined as the time to death or the use of permanent assisted ventilation (tracheostomy or ventilatory support for ≥16 h/day for >21 continuous days in the absence of an acute reversible event).18
At baseline, infants in the nusinersen group had earlier onset of symptoms and greater burden of disease compared with the control group. A pre-specified interim analysis at 2 years showed a significantly higher percentage of infants treated with nusinersen had a motor milestone response (41% versus 0% controls, p<0.001). These results prompted early termination of the trial. In the final analysis, 51% of the infants in the nusinersen group and no infants in the control group had a motor milestone response. At final analysis, 39% of the infants in the nusinersen group and 68% in the control group had died or received permanent assisted ventilation. The median time to death or permanent assisted ventilation was 22.6 weeks in the control group and was not reached in the nusinersen group. Overall, the risk of death or the use of permanent assisted ventilation was 47% lower in the nusinersen group than in the control group (p=0.005).17 Based on these results, on 23 December 2016, the US Food and Drug Administration (FDA) approved Spinraza® (nusinersen; Biogen, Durham, NC, USA) for all ages.20
Risdiplam is an oral, small molecule, pre-mRNA splicing modifier that increases the production of the SMN protein, and in that way is related to the current discussion of nusinersen.19,21–24 Its efficacy results from its unique SMN2 pre-mRNA binding sites: a 5’ splice site in intron 7 and exonic splicing enhancer in exon 7. This increases levels of full-length SMN mRNA and protein. Risdiplam has a significant advantage over nusinersen since it is the only oral medication approved for SMA,25 though its oral dosing has potentially more advantages than just ease of administration. Upon absorption it will reach and be expressed in extraneuronal tissues where SMN protein is known to be deficient.26 Such tissues include skeletal muscle, heart, bone, and autonomic and other nervous systems that may be contributing to disease state.27 On 7 August 2020, the FDA approved Evrysdi® (risdiplam; Genentech, San Francisco, CA, USA) to treat patients 2 months and older with SMA. There is limited space in this review to fully discuss this compound but efficacy and safety have been established in a series of clinical trials.19,21–24
The application of risdiplam in the clinic will be further commented on in the Conclusions.
Gene replacement therapy
The path to gene replacement therapy for SMA was developed at the Nationwide Children’s Hospital in Columbus, OH, USA. Two key factors enabled clinical trials in this area: the SMNΔ7 mouse model was available, and adeno-associated virus serotype 9 (AAV9) was shown to target neonatal neurons following intravascular delivery.28 Preclinical studies demonstrated that self-complementary (sc)AAV9 infused on post-natal Day 1 rescued SMNΔ7 pups. Survival was increased from 15.0 days to 28.5 days with vector dosing of 6.7×1013 vg/kg, and to more than 250.0 days with doses of 3.3×1014 vg/kg. Treatment on post-natal Day 5 showed only partial correction, and post-natal Day 10 had little effect.29
Pilot single-centre phase I/II clinical trial in spinal muscular atrophy type 1
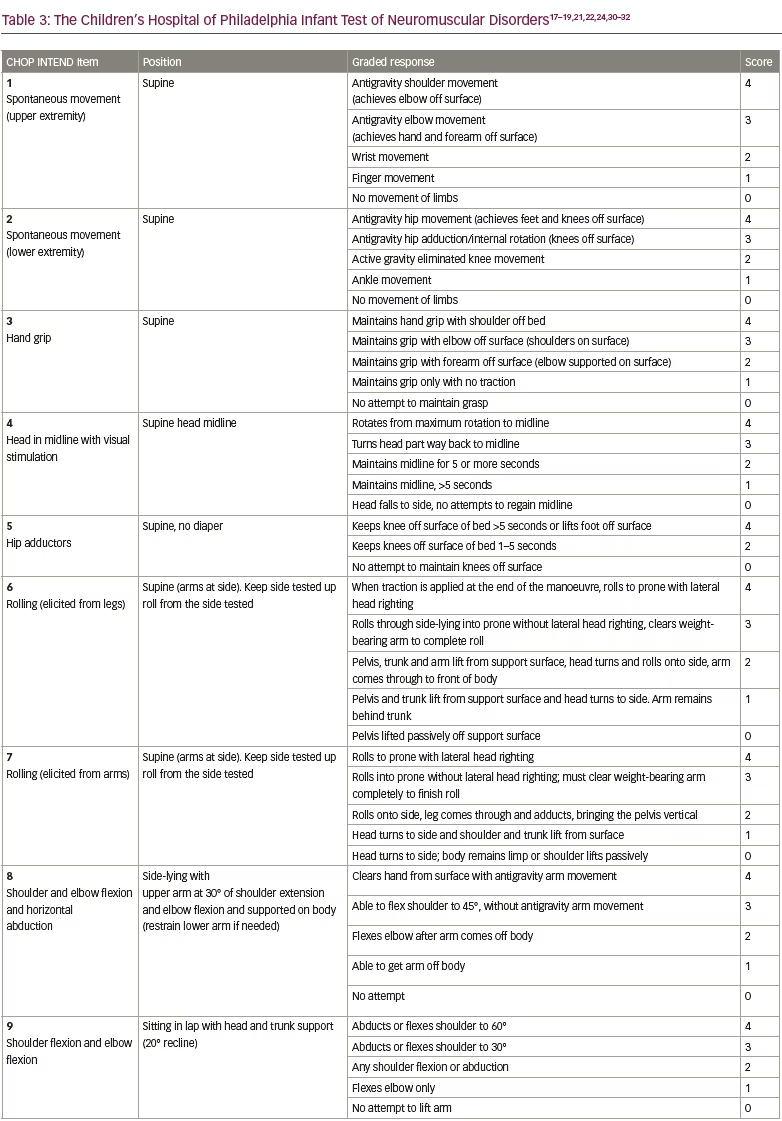
Preclinical data defined the protocol for the first clinical trial, now named ‘START’, which started enrolling in 2014.30 The treatment paradigm, as performed in the clinic, is shown in Figure 2. This was a dose-escalation trial in which patients were enrolled in two cohorts according to the dose administered. The primary outcome was safety, and the secondary outcome was time until death or the need for permanent ventilatory assistance. Exploratory outcomes included motor milestone achievements (particularly, sitting unassisted) and scores from Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND; scores patients according to motor skills on 16 items; Table 3).17–19,21,22,24,30–32
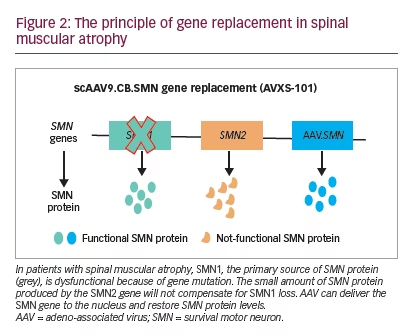
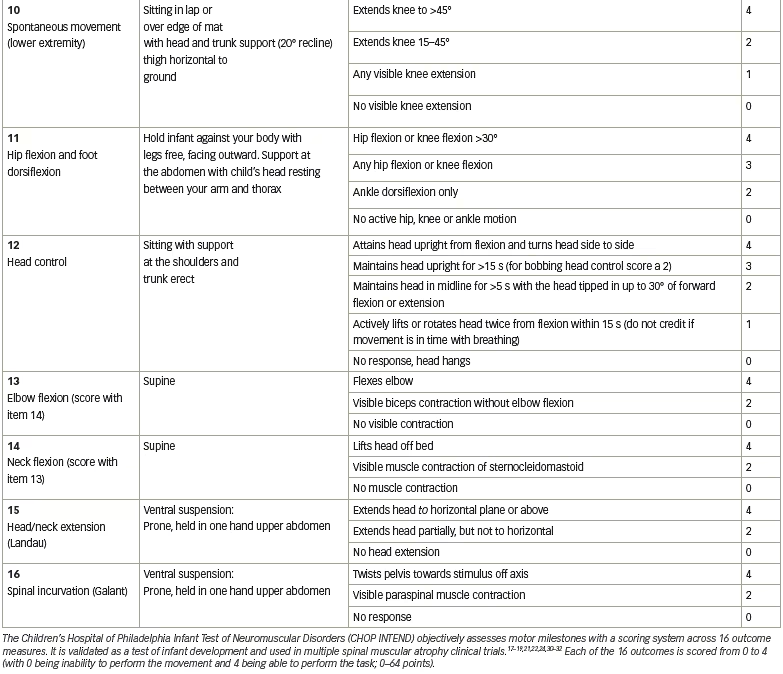
Fifteen patients were enrolled in the clinical trial planned for infants from newborns to 9 months of age. Of the 16 patients screened, one was excluded because of elevated AAV9 antibody titres >1:50. Cohort 1 (n=3, mean age 6.3 months) received a single intravenous dose of scAAV9.CB.SMN (AVXS-101), 6.7×1013 vg/kg body weight. The first patient developed elevated serum aminotransferases (alanine aminotransferase 31 × upper limit of normal [ULN]; aspartate aminotransferase 14 × ULN) meeting criteria for a serious adverse event (SAE; Figure 3).30,33 Consequently, the protocol was amended, with all subsequent patients treated with oral prednisolone 1 mg/kg/day for 30 days, starting 24 hours before gene delivery. The amendment also included a dose adjustment for Cohort 2 (n=12; mean age 3.4 months), which received a single-dose vector 2.0×1014 vg/kg – half that used in preclinical trials.30 Such was the impact of these protocol amendment, that this starting dose, alongside prednisone as an immunosuppressant, has been used in most subsequent neuromuscular clinical gene therapy trials.34
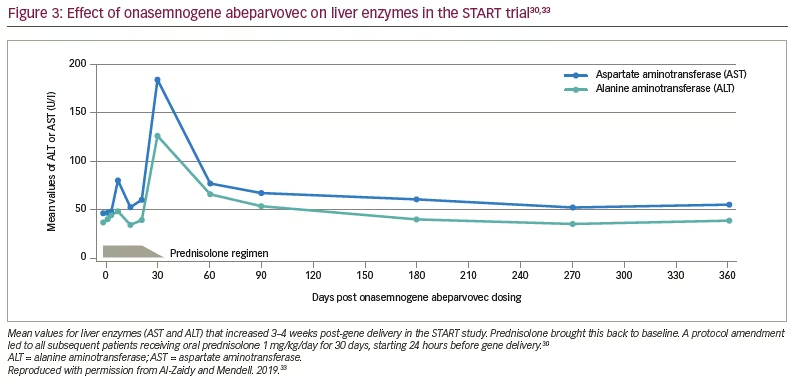
As of 7 August 2017, all the patients had reached age ≥20 months without needing permanent mechanical ventilation (historical controls showed >90% of patients were dependent on permanent mechanical ventilation or had died at this age).30,35 Eleven patients were able to speak, a milestone rarely achieved in infants with SMA type 1. All the patients had increased CHOP INTEND scores from baseline and maintained these changes during the trial (Figure 4).30 Cohort 2 had a mean increase of 9.8 points at 1 month and 15.4 points at 3 months. Sitting was achieved in 11 of 12 patients in Cohort 2, and nine could sit for at least 30 seconds. Most gratifying was the clinically meaningful results of feeding (hand to mouth), talking and sitting for at least 30 seconds in 11 of 12 patients in Cohort 2.27
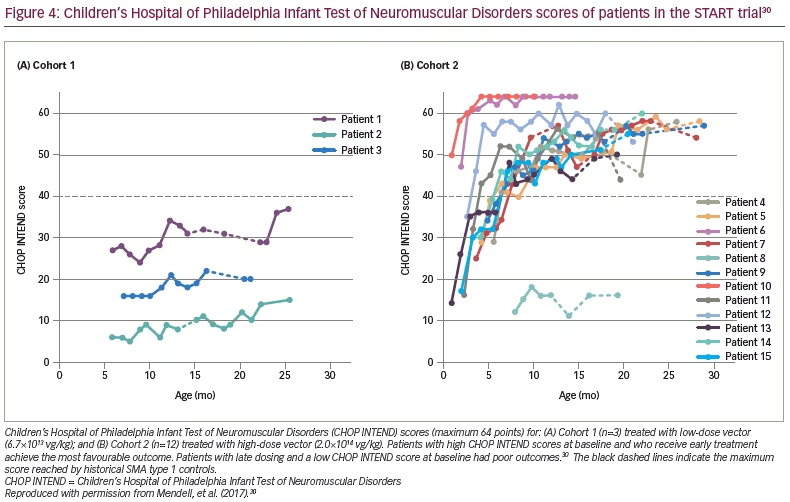
Two findings were related to age at gene delivery. The oldest patient treated was 7.9 months, and achieved little or no measurable benefits, thus, defining the limits of efficacy. On the other hand, the two youngest patients, who had the highest CHOP INTEND scores at the time of enrollment, achieved the highest scores on CHOP INTEND (>60 points) at the conclusion of the trial, and were able to crawl, stand and walk (Figure 4).30 Both of these patients’ families were alerted to SMA prior to delivery because of family history.30 These outcomes strongly encourage early treatment and newborn screening for SMA.
The START trial was groundbreaking and defied all predictions about safety and efficacy issues for gene therapy. All 15 patients surpassed the previously reported median age of survival,35 and motor milestones and clinically meaningful outcomes never before seen were achieved in this clinical trial.30 As a result, the FDA approved onasemnogene abeparvovec gene therapy for SMA on 24 May 2019, under the commercial name Zolgensma® (Novartis, Durham, NC, USA). Multiple important follow-up clinical trials have been sponsored by Novartis, adding to the use of onasemnogene abeparvovec.31,36
Phase III trial confirming efficacy and safety in spinal muscular atrophy type 1
A confirmatory, 12 centre, open-label, phase III trial (STR1VE) followed.36 Eligibility criteria included patients aged ≤6 months with one or two copies of SMN2. Like START, this was a single intravenous dose of onasemnogene abeparvovec at a dose equivalent titre (1.1×1014 vg/kg). Co-primary efficacy endpoints were independent sitting for ≥30 seconds per Bayley-III at 18 months and survival (absence of death or permanent ventilation) at age 14 months. Historical controls from the Pediatric Neuromuscular Clinical Research dataset (PNCR) represented the comparator group.36
Twenty-five patients were screened for AAV9 antibodies, and three were excluded with higher titres. The mean age at gene transfer was 3.7 months (12 females and 10 males), and none required feeding or ventilatory support. Thirteen (59.1%) patients achieved independent sitting ≥30 seconds at 18 months versus none in the control group (p<0.0001). At age 18 months, 18 patients did not use ventilatory support versus no PNCR controls (p<0.0001). Of the seven patients using non-invasive ventilation, five had documented previous use of Trilogy 100 and two had used other types of non-invasive ventilation. In addition, a clinically meaningful measure of ability to thrive at 18 months included three assessments: (1) ability to tolerate thin liquids by clinical swallowing assessment (55%); (2) able to feed exclusively by mouth (86%); and (3) weight maintained and consistent with age (14%).36
Regarding CHOP INTEND scores, there was a mean increase from baseline of 6.9 points at 1 month post-dosing, 11.7 points at 3 months and 14.6 points at 6 months.36 Twenty-one (95%) patients achieved a CHOP INTEND score of ≥40.0 points, 14 (64%) achieved ≥50.0 points and five (23%) achieved ≥60.0 points. Historically, children with SMA type 1 almost never achieve CHOP INTEND scores >40 points.35
The safety profile demonstrated in STR1VE was again remarkable and convincing for continued use of onasemnogene abeparvovec. All adverse events were documented, but only three (14%) SAEs were related to gene delivery. Two of these SAEs were elevated liver enzymes, a finding consistent with other AAV clinical trials. The third SAE was hydrocephalus, occurring for unknown reasons. This trial validated the findings of the START trial and opened avenues for gene therapy for many other childhood diseases.
Gene therapy for spinal muscular atrophy type 2
Considering the success of onasemnogene abeparvovec in treating infants with SMA type 1, on-going studies are addressing its potential in treating patients with sitting, non-ambulatory SMA type 2, and SMA type 3. Treatment at an older age would require higher intravenous dosing of onasemnogene abeparvovec due to body weight, accompanied by a higher risk of side effects. To circumvent these obstacles, gene transfer directly targeting the bulbar and spinal cord motor neurons was planned by intrathecal delivery. Preclinical studies showed that intrathecal delivery to mice and non-human primates was safe and effective at a viral vector tenfold lower than with IV dosing.37,38
The phase I, open-label, ascending-dose STRONG study started in December 2017.39 Patients with SMA type 2, aged between 6 and <12 months, with three copies of SMN2, who were able to sit unassisted for 10 seconds but unable to walk or stand were included. Those with severe scoliosis (≥50° curve) were excluded. There were three onasemnogene abeparvovec dosing cohorts: Cohort 1, receiving 6.0×1013 vg; Cohort 2, receiving 1.2×1014 vg; and Cohort 3, receiving 2.4×1014 vg. Each group was stratified by age, either 6 to <24 months and 24 to <60 months. The primary endpoints were safety/tolerability, independent standing for ≥3 seconds in patients aged 6 to <24 months or change in Hammersmith Functional Motor Scale–Expanded score in patients aged 24 to <60 months. Outcomes were compared with those of PNCR historical controls. Patients received prophylactic prednisolone (1 mg/kg/day) 24 hours prior to intrathecal delivery, maintained for approximately 30 days with a taper depending on clinical toxicity. Onasemnogene abeparvovec was delivered as a single intrathecal injection, with the patient sedated and in the Trendelenburg position at 30° to enhance distribution to the spinal cord and brain.
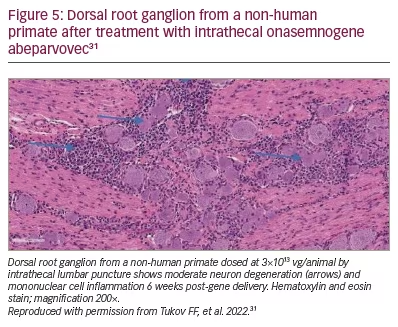
In October 2019, as enrolment was complete for Cohort 1 (n=3) and Cohort 2 (n=25) but incomplete for Cohort 3 (n=4), the FDA imposed a partial clinical hold on the study resulting from a Novartis report to the FDA of unexpected findings in the dorsal root ganglion (DRG) in onasemnogene abeparvovec-treated non-human primates.40 This preclinical study showed mononuclear cell inflammation, sometimes accompanied by neuronal cell body degeneration or loss (Figure 5), though the non-human primates were asymptomatic.38 Novartis then did further research to assess clinical impact. Intrathecal delivery of vector in additional studies showed DRG and trigeminal ganglion inflammation with scattered neuronal degeneration. The findings at 52 weeks post-dosing were non-progressive and were of minimal severity compared to interim autopsy data performed at earlier time points.38 In August 2021, the FDA determined that the STRONG study could proceed with intrathecal delivery.41
Despite release from clinical hold, the sponsor elected not to enrol more patients. Safety issues appeared minimal following intrathecal delivery and the SAEs related only to transaminase elevations without increase in bilirubin. No signs of DRG toxicity were encountered.39
Further studies are needed to validate the efficacy of intrathecal delivery in SMA type 2. To address this, Novartis is sponsoring STEER, a randomized, sham-controlled, double-blind phase III study (Efficacy and safety of intrathecal OAV101 [AVXS-101] in pediatric patients with type 2 spinal muscular atrophy (SMA); ClinicalTrials.gov identifier: NCT05089656).42 In the STEER study, treatment-naïve patients with SMA type 2, aged 2–17 years, will be treated with intrathecal onasemnogene abeparvovec at 1.2×1014 vg, a safe dose used in the STRONG trial.
Impact of gene therapy for newborn infants with spinal muscular atrophy
SMA was added to the Department of Health and Human Services’ Recommended Uniform Screening Panel in February 2018, a little more than a year after nusinersen was approved for SMA treatment and three months after the START study in SMA type 1 gene therapy was published.30,43 Currently 48 states include SMA in their newborn screening panel (Figure 6).44
The value of onasemnogene abeparvovec is unequivocal for treating newborns, evidenced by a phase III, multicentre, single-arm trial (SPR1NT).31,32 SPR1NT included infants with pre-symptomatic SMA, with biallelic SMN1 deletions, and two or three copies of SMN2. Other newborns were excluded because of signs of SMA, reduced compound muscle action potentials and elevated AAV9 antibody titres. Infants with two copies were treated at median age of 21 days (range 8–34 days), and those with three copies were treated at median age of 32 days (range 9–43); the first was enrolled in April 2018.31,32
For the cohort of 14 infants with two copies of SMN2, the efficacy and safety of onasemnogene abeparvovec was demonstrated, and outcomes were remarkably consistent given that all 14 patients achieved the primary endpoint of independent sitting for at least 30 seconds (p<0.0001). All 14 (100%) achieved motor milestones assessed by Bayley-III, and 11 of 14 stood alone and walked independently. All children in this cohort of the SPR1NT trial achieved a CHOP INTEND score of at least 58 by 18 months of age. None required any form of respiratory or feeding support.31
Results from the second cohort of patients, those with three copies of SMN2, are important in translating the findings into clinical practice because most infants with this molecular profile will develop clinical manifestations consistent with SMA type 2 or 3. Three copies of SMN2 predicts onset of SMA type 2 at age 7–18 months in 54% of patients, and type 3 phenotype in 31%. Patients with SMA type 2 can sit independently, some can stand and none can walk.32
For the cohort of infants with three copies of SMN2, 15 infants (13 identified by newborn screening) were analysed. All 15 patients were able to stand independently for at least 3 seconds at a median age of 377 days, 14 patients walked independently by 24 months of age, and all patients had Bayley Scales of Infant and Toddler Development motor scores comparable to neurologically ‘normal’ infants. All survived, without respiratory or nutritional support by trial end.32
The safety profile of early intravenous treatment was exceptional in both cohorts of patients in SPR1NT. In patients with two copies, mild hepatoxicity was observed in only three subjects. Troponin-I elevation was rarely encountered and there was no compromised cardiac function by echocardiogram. Platelet counts remained >75,000 for all infants throughout the study.31,32
Commercial gene transfer treatment of 21 infants from Ohio confirmed the lessons learned from SPR1NT.45 Infants who were recognized by newborn screening and were treated early faired the best. Children older than 6 months were more likely to have asymptomatic transaminase elevation and were treated with longer immunosuppression.45 The safety information for Zolgensma® is summarized in Table 4, including reports on recent deaths.46,47

Conclusion
The results of molecular-based treatment for SMA type 1, when used prior to onset of symptoms, are overwhelmingly successful for a disease predicting death by age 2 years. Treatment outcomes in infants with SMA type 1 vary depending on age and severity of disease at gene transfer. The FDA has approved intravenous systemic gene delivery for patients up to 2 years of age, and for pre-symptomatic infants, we have observed sustained benefit for nearly 5 years.48 Results of treatment of SMA type 2 require further evaluation.
For patients still showing manifestations post-gene delivery, it is acceptable to consider combination treatment with other approved therapies for SMA (nusinersen or risdiplam). There are no clinical trials directly addressing this question, but favourable results have been reported with gene transfer and nusinersen without an increase in adverse events.49,50 Thus, patients receiving gene therapy beyond 6 months of age with residual signs of disease following treatment might benefit by receiving nusinersen. Similar studies are not currently available with risdiplam.
In patients for whom first-line gene therapy is not possible (e.g. those with pre-existing AAV antibody), the choice between nusinersen and risdiplam may be difficult for physicians. There are no head-to-head trials comparing efficacy, but overall, the results support the use of risdiplam as an important alternative to nusinersen for the treatment of patients with SMA type 1.51
SMA gene therapy has been a leading example of safety and efficacy of gene therapy, especially for other infant genetic diseases. As a breakthrough treatment, it provides a path for gene delivery for older children and adults.