Amyotrophic lateral sclerosis (ALS) is characterized by the degeneration of both upper and lower motor neurons, which ultimately leads to muscle weakness, atrophy, spasticity and contractures.1 ALS typically manifests in the 50–60 years age range, although familial cases may present in late adolescence or early adulthood.2 The time from the first symptom to diagnosis is approximately 10–16 months, underscoring the difficulty of timely initiation of therapeutic intervention at an early stage.3 The relentless progression of the disease eventually leads to respiratory muscle dysfunction and limits survival to 3–5 years after the onset of the disease.4
Approximately 10% of ALS cases are hereditary with an autosomal dominant pattern of inheritance, and the known genes associated with ALS, including SOD1, C9ORF72, FUS and TARDBP, collectively account for approximately 40–55% of all familial forms.5,6 In fact, approximately 50 genes that influence the course of ALS have been identified.4,6 In total, 90% of cases are sporadic and do not present with a familial history. The aetiology of these instances remains undetermined in the majority of cases.7 The incidence of sporadic ALS (sALS) is greater in men than in women, with a ratio of 2:1, whereas the incidence of familial ALS (fALS) is comparable between the sexes.8
The epidemiology of ALS involves variations in disease prevalence, which are influenced by genetic inheritance factors, leading to differences in morbidity levels across various regions. For example, in Europe and North America, where the population primarily has European ancestry, the incidence of ALS is somewhat higher than the global average, ranging from 1.71 to 1.89 per 100,000 people per year.9 These findings suggest that genetic or environmental factors specific to these populations may contribute to a higher frequency of ALS occurrence. In contrast, Asian countries, particularly South Asia and West Asia, have lower ALS incidence rates, with frequencies ranging from 0.73 to 0.94 cases per 100,000 people per year.10 Oceania, which includes countries such as Australia and New Zealand, has some of the highest ALS incidence rates in the world, at 4.42 per 100,000 people per year, indicating a pronounced prevalence of the disease in this region, which may be linked to a combination of genetic predispositions and environmental factors unique to Oceania.10,11
Aetiology and pathogenesis
The onset and progression of ALS are facilitated by multiple factors. Genetic and phenotypic differences among patients complicate the understanding of the pathogenetic mechanisms of ALS, which involve a multitude of genes and cellular processes, including disturbances in RNA metabolism and protein homeostasis, defects in nucleocytoplasmic transport, DNA repair impairments, excitotoxicity and oxidative stress. Research has also shown that the development of ALS is determined by a complex interplay of genetic factors and environmental influences.12
Environmental factors
Environmental factors play a significant role in the etiopathogenesis of ALS (Figure 1). A study conducted by a group of scientists led by Newell in 2022 is particularly notable for its in-depth analysis of the impact of environmental factors on population health. Their research revealed a direct link between specific environmental factors and an increased risk of developing ALS. Specifically, substances such as β-N-methylamino-L-alanine (BMAA), with an odds ratio (OR) of 2.32, formaldehyde (OR=1.54), heavy metals overall (OR=2.99), manganese (OR=3.85), mercury (OR=2.74) and zinc (OR=2.78) significantly increase the likelihood of developing ALS.13 However, additions to this list were proposed later in a study by Zhu et al. in 2023, indicating that factors such as head injuries (OR=1.26), physical activity (OR=1.06), electric shock (OR=2.72), military service (OR=1.34), pesticides (OR=1.96) and lead (OR=2.31) can also contribute to the development of ALS.12 Moreover, factors such as cerebrovascular diseases (OR=0.99), agricultural and industrial conditions (OR=1.22 and 1.24, respectively), smoking (OR=1.25) and heavy metals overall (OR=1.5) were not recognized as significant risk factors according to the results of this meta-analysis. Notably, electric shock injury has an ambiguous interpretation because data in the literature indicate a lack of association with the development of ALS.14,15 Interestingly, type 2 diabetes (OR=0.74) was identified as a factor that reduces the risk of developing ALS.12 Further research by Duan et al. in 2023 confirmed the importance of pesticides (OR=1.46), past head injuries (OR=1.37) and military service (OR=1.29) as risk factors, supplemented by the effects of magnetic fields (OR=1.22), solvents (OR=1.37) and hypertensive disease (OR=1.04). Moreover, from their perspective, heavy metals (OR=1.79) and cerebrovascular diseases (OR=1.26) are still risk predictors. Additionally, the intake of antidiabetic drugs (OR=0.52), a high body mass index (OR=0.60 in obese individuals and overweight individuals compared with normal individuals and underweight individuals), urban living conditions (OR=0.70), diabetes (OR=0.83) and kidney diseases (OR=0.84) may reduce the risk of developing ALS.16
Figure 1: Data categorized on the basis of their potential impact on the risk of developing amyotrophic lateral sclerosis
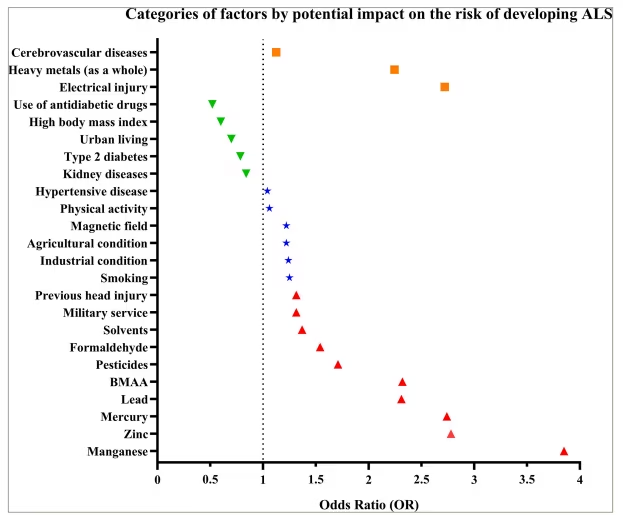
Each point represents the odds ratio (OR) for a specific factor: the red upper triangles indicate factors that increase the risk of developing ALS; the green lower triangles represent protective factors that potentially reduce the risk; the blue stars represent factors that can contribute to ALS risk; and the orange squares represent factors with conflicting data. The vertical dotted line (OR=1) visually separates factors by their potential impact on risk: the values above this line indicate increased risk or no significant impact, whereas those below the line indicate a protective effect.
ALS = amyotrophic lateral sclerosis; BMAA = β-N-methylamino-L-alanine.
According to the provided diagram, BMAA, manganese, mercury, zinc and lead are the most significant environmental factors of causality in ALS. According to the literature, BMAA causes damage to motor neurons through the activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate and mGLuR5 receptors and affects the activity of the cystine/glutamate antiporter (SxC-), enhancing excitotoxicity and oxidative stress, which contributes to neuronal damage.17,18 Under natural conditions, humans may be exposed to BMAA, which is present in drinking and recreational waters, as well as through the consumption of food sources such as aquatic and terrestrial fauna, edible flora and dietary supplements derived from cyanobacteria.19
Manganese can reduce adenosine triphosphate (ATP) levels, signalling through P2 receptors, and decrease insulin/insulin-like growth factor 1; it also affects the PI3K/Akt and mitogen-activated protein kinase signalling pathways. It can also act through neuroinflammation via the cGAS-STING, NLRP3-CASP1, NF-κB, SIRT and JAK/STAT signalling pathways, promoting motor neuron degeneration.20 Exposure to manganese can occur through various pathways, including environmental sources, occupational environments, dietary intake, total parenteral nutrition, abuse of the drug methcathinone or genetic predispositions, such as mutations in the SLC30A10 transporter gene.21
Mercury is known to induce direct and indirect post-translational modifications of DNA, such as phosphorylation, ubiquitination, acetylation, nitrosylation and S-mercuration of target proteins involved in many cellular processes. Polymorphisms in genes associated with glutathione, mercury transport proteins (e.g. metallothioneins or ATP-binding cassette transporters), cytochrome p450 3A, ε4 APOE and brain-derived neurotrophic factor (BDNF) are also linked to the toxic effects of mercury on biological tissues. The induction of oxidative stress, which activates the process of mitochondrial autophagy, is the initial stage of mercury neurotoxicity, including the development of ALS.22 Regular exposure to mercury may arise from a multitude of sources. The diet represents a major route, particularly through the consumption of fish and seafood, which are notable for their content of methylmercury. This compound is subsequently metabolized by mammals into inorganic mercury to some extent. Furthermore, meat and poultry may also contain inorganic mercury if the feed given to the animals includes fish-based components. In some developing regions, medicinal and cosmetic products, such as skin-whitening creams, incorporate mercury. Occupational exposure risks include inhalation of mercury vapour and potential contamination through workers’ clothing.23
Zinc is capable of selectively inhibiting BDNF, which, as mentioned earlier, is also one of the mechanisms of neurotoxicity.24 Zinc can also contribute to stress mediated by the endoplasmic reticulum (ER), which, through specific interactions with Derlin-1, a component of the ER-associated degradation mechanism, leads to subsequent motor neuron death.25 This leads to the aggregation and accumulation of the TDP-43 protein in the cytoplasm.26 The likelihood of excessive zinc consumption is increased by the utilization of dietary supplements or the ingestion of foods fortified with zinc beyond advised thresholds. Workers engaged in sectors such as welding, zinc mining and smelting are particularly susceptible to exposure to zinc dust or fumes, increasing their risk of zinc toxicity. Additionally, environmental contributors, such as zinc-contaminated water, can inadvertently increase zinc exposure.27
Notably, the variability of ORs for identical factors across different studies may be associated with a number of factors. These include the use of samples that differ in the demographic characteristics of participants, such as age, gender and ethnic background. The size of samples also plays a significant role: larger samples help increase statistical significance and refine results, whereas studies with smaller samples may show a wider range of OR estimates. Furthermore, the design of the study, strategies for accounting for the influence of external variables, the diversity of approaches to defining and quantitatively assessing exposure factors and the chosen statistical methodologies can significantly affect the OR estimates.
Thus, the presented data indicate a significant link between exposure to a number of environmental agents and an increased risk of developing ALS, underscoring the importance of understanding the environmental risk factors for ALS and the need to develop strategies to reduce these exposures among the at-risk population.
External factors can influence gene activity without directly changing the DNA sequence through processes such as DNA methylation, histone modification and the impact of noncoding RNAs, including microRNAs. These findings indicate that the interaction between the environment and genes in the development of ALS highlights the critical role of epigenetic changes.28
Genetic factors
As mentioned earlier, approximately 50 genes that are potentially associated with a high risk of developing ALS have recently been identified.4,6 Notably, four genes account for up to 70% of all fALS cases, namely, SOD1, TARDBP (TDP-43), C9orf72 and FUS.29 Disease-causing variants in the nucleotide sequences of other genes occur relatively rarely.30
A missense variant (the most common variants being D90A, A4V and G93A) in the SOD1 was the first genetic cause of ALS described by Rosen et al. in 1993.31,32 This type of nucleotide sequence variant leads to instability of the mutant protein, which underlies 12–20% of fALS cases and 1–2% of patients with sALS.33,34
Interestingly, ALS does not arise as a result of SOD1 deactivation. Animal studies have shown that knocking out this gene does not necessarily lead to symptoms of ALS. This finding suggests that while variants in SOD1 are associated with some cases of fALS, the absence of SOD1 enzyme activity itself does not cause the disease. It is likely that pathological changes are linked not to the loss of its normal function but to toxic effects arising from functional changes in the mutated gene.35,36 However, the deactivation of SOD1 does not have consequences and indeed leads to outcomes such as the development of progressive distal motor axonopathy.37
Nucleotide sequence variants induce structural and functional changes in SOD1, which, through various mechanisms, lead to their toxic effects. A distinctive feature of this point variant is the formation of insoluble ubiquitin-positive inclusion bodies in motor neurons, which are a key hallmark of ALS pathology.38
Importantly, heat shock proteins play crucial roles in maintaining protein homeostasis by assisting proteins in proper folding or facilitating the degradation of improperly folded proteins. In the case of disrupted misfolded and aggregated proteins, HSP70 binds to the hydrophobic patches exposed on the outside (under normal protein folding, such patches are turned inward of the molecule) and, through the recruitment of ubiquitin ligase, directs the protein for degradation in the proteasome.39 In the case of ALS, chaperones become encapsulated in mutant SOD1, which disrupts their apoptotic activity.40 This leads to a reduced ability of cells to cope with improper protein folding, thereby contributing to the development and progression of the disease.
Notably, the accumulation of abnormal protein aggregates can cause ER stress, which promotes the activation of the ubiquitin‒proteasome system (UPS), creating a positive feedback loop that ultimately leads to the exacerbation of cellular dysfunction.41
Considering the data presented here, a new class of antisense oligonucleotides, named Tofersen, received accelerated approval from the US Food and Drug Administration (FDA) in April 2023 for the treatment of ALS caused by the SOD1 variant. This medicinal product consists of short, synthetic, single-stranded RNA or DNA molecules that bind to a complementary sequence and alter mRNA expression.42 Tofersen directly binds to and facilitates the degradation of SOD1 mRNA produced by mutated SOD1, effectively leading to a reduction in SOD1 protein synthesis.43,44 From a clinical standpoint, the intrathecal administration of Tofersen in patients with SOD1-linked ALS has shown significant efficacy. This treatment reduces markers of neurodegeneration, notably both serum neurofilament light chain (NfL) and phosphorylated neurofilament heavy chain in the cerebrospinal fluid (CSF), and decelerates the rate of functional deterioration. Furthermore, Tofersen sustains patient quality of life and possesses a manageable safety profile. However, vigilant monitoring of autoimmune responses is essential to ensure patient safety and optimize therapeutic outcomes.45
Interestingly, in a study conducted by Forsberg et al. in 2019, granular SOD1-immunoreactive inclusions were found in the motor neurons of patients with ALS who did not have typical variants in the SOD1 sequence. These findings suggest that the abnormal structure of the mutant SOD1 protein could be the result of a more complex pathogenetic process induced by nucleotide sequence variants in a range of other genes, such as C9ORF72HRE, FUS, KIF5A, NEK1, VAPB and ALSIN. These details imply the presence of complex interactions between various genetic elements in the development of ALS, which can occur even in the absence of direct variants in SOD1.46
The protein TDP-43 is encoded by TARDBP and is involved in the regulation of gene transcription and RNA processing.47 Nucleotide sequence variants (predominantly missense variants) in TARDBP can lead to a specific form of neurotoxicity, which is an important pathogenetic factor in the development of ALS.48 These variants affect the function of TDP-43, potentially causing its misfolding and aggregation in motor neurons, leading to accumulation due to the inhibition of activity and damage to the UPS. The exact mechanisms by which TDP-43 causes damage are not fully understood. In 2021, Yin et al. demonstrated that TDP-43 interacts with specific proteins (e.g. PAC2 [a chaperone involved in the maturation of the 20S proteasome subunit] and PSD-95 [postsynaptic density protein 95]) and pathways (e.g. the autophagolysosomal system), disrupting the function of the UPS, which impedes the normal breakdown of proteins, thereby leading to cellular dysfunction and death.49
Importantly, most patients with ALS, including those with sALS without pathogenic nucleotide sequence variants in TARDBP, as well as individuals with hexanucleotide repeat expansions in C9ORF72, also accumulate cytoplasmic TDP-43 protein aggregates, leading to the formation of inclusion bodies and resulting in cellular dysfunction. Moreover, the reduction in TDP-43 levels in the nucleus, due to its redistribution to the cytoplasm, can disrupt the regulation of mRNA metabolism and lead to cellular dysfunction.50–52
In 2011, DeJesus-Hernandez et al. reported that one of the genetic causes of ALS is associated with a specific variant of C9ORF72 in the form of a hexanucleotide repeat expansion, GGGGCC, also referred to as G4C2. In healthy individuals, this GGGGCC sequence repeats fewer than 30 times within an intron of C9ORF72. However, in people with ALS, this sequence may repeat hundreds or even thousands of times, leading to a loss of normal function.53 The aggregated prevalence of C9ORF7 repeat expansion among patients with fALS stands at 23% (confidence interval [CI]: 18–28%). In contrast, the prevalence in patients with sALS is reported to be 3% (CI: 3–4%).54
There is substantial evidence affirming a direct causal association between C9ORF72 expansion and frontotemporal dementia (FTD).54,55 The mutation frequency of C9ORF72 in familial cases of FTD is approximately 20%.54 Clinically, the most common phenotype of FTD associated with the C9ORF72 mutation is the behavioural variant FTD, which frequently presents concomitantly with the characteristics of ALS.55
The presence of this nucleotide sequence variant leads to toxic effects in the form of RNA amplification, aggregation of proteins with dipeptide repeats and haploinsufficiency of C9ORF72.56 Additionally, due to the presence of the C9ORF72 variant, disruptions in the function of the UPS occur, leading to the appearance of ubiquitin-positive inclusion bodies.57 Notably, individuals with nucleotide sequence variants in C9ORF72 exhibit a significant reduction in the number of proteasome subunits, leading to the formation of abnormal forms that are present in cytoplasmic inclusion bodies. The presence of these proteins in these aggregates indicates that proteasomes cease to function efficiently, ultimately leading to the accumulation of misfolded proteins and cellular dysfunction.58
It is hypothesized that the C9ORF72 gene also plays a specific role in the initiation and regulation of autophagy. According to the data obtained, the downregulation of C9ORF72 due to genetic variants has a negative effect on the initiation phase of autophagy, disrupting normal cellular clearance processes.57
The list of possible RNA processing disorders includes phenomena such as abortive transcription, problems with splicing introns containing G4C2 sequences and aggregation phenomena in the cell nucleus.59 Additionally, RAN translation has been identified for a portion of transcripts with G4C2 repeats, leading to the synthesis of pathological dipeptides, which form inclusions in neurons, potentially contributing to the development of neurodegenerative processes.60 Furthermore, nuclear structures that disrupt the function of RNA-binding proteins can form from RNA transcripts with G4C2 repeats. These structures directly affect gene expression and the RNA splicing process.61
The FUS protein, a member of the RNA-binding protein family, plays a key role in RNA metabolism and DNA repair processes. Predominantly localized in the nucleus, it regulates transcription, pre-mRNA splicing, RNA transport and the stability of RNA.62 Currently, approximately 50 different nucleotide sequence variants of FUS (predominantly missense variants) have been identified in patients with fALS, which leads to disruptions in nucleocytoplasmic transport and the redistribution of the FUS protein to the cytoplasm, subsequently leading to the formation of immunoreactive inclusions, along with the direct toxic impact of the soluble form of FUS. Changes in the ratio between the nuclear and cytoplasmic contents also affect the ability of proteins to adequately perform their functions in the nucleus.63,64 Therefore, both the loss of nuclear function and increased cytoplasmic cytotoxicity of FUS contribute to the pathogenesis of ALS.
Notably, despite significant progress in identifying genes associated with ALS, much remains unknown about the genetic architecture of this disease. Research suggests the possibility of an oligogenic origin of ALS, where combinations of different genetic variants may increase susceptibility to the disease and accelerate its progression. The role of pleiotropy is also discussed, where one gene can cause several different phenotypic effects. In exploring the genetic complexity of ALS, the pleiotropic effects of specific gene mutations, such as those found in SOD1, offer profound insights into the diverse clinical presentations of this disease. Notably, the p.I114T mutation in SOD1 exemplifies how a single genetic alteration can influence multiple phenotypic traits in patients with ALS. Research has demonstrated that this mutation is significantly over-represented among Australian sALS cases compared with controls, suggesting a robust association with the disease. Individuals carrying this mutation experience a range of clinical outcomes, including variations in the age of onset, progression rates, and overall survival. These differences highlight the pleiotropic nature of the mutation. This case underscores the intricate genetic architecture of ALS and emphasizes the need for a nuanced understanding of its genetic contributions to neurodegenerative diseases.65
Disruptions of proteostasis
The complex molecular landscape of ALS unfolds due to disruptions in protein homeostasis, which is a crucial aspect of this pathology. Protein aggregates are clusters of misfolded proteins that accumulate inside cells, resulting from an imbalance between synthesis and degradation processes. In ALS, these protein aggregates are typically found in the cytoplasm of motor neurons. Under normal circumstances, these proteins are predominantly localized in the nucleus, and their presence in the cytoplasm indicates cellular dysfunction.66,67 In the context of ALS, these aggregates mainly consist of proteins such as TDP-43, neurofilaments, FUS, SOD1 and tau protein.68–75 Their aggregation is a common pathological feature observed in the neurons and skeletal muscle cells of patients with ALS. For example, TDP-43 aggregates are found in 98% of both sALS and fALS cases, emphasizing their central role in the disease mechanism.69 SOD1 is associated with reduced expression of UPS components, and transitional ER ATPase (TER ATPase or valosin-containing protein) and ubiquilin-2 play significant roles in substrate delivery to the proteasome, which is disrupted by nucleotide sequence variants in ALS-related genes.76 Dysregulation of chaperone proteins manifests with SOD1 and TARDBP variants.68 Protein aggregation leads to disruptions in proteostasis within the cell, causing stress. This process can isolate essential RNAs and proteins, interfere with normal axonal transport and hinder protein breakdown, especially their degradation via the ubiquitin-dependent pathway. It is hypothesized that the energy depletion of motor neurons is a result of the costly metabolism of misfolded proteins.77
In addition to these molecular insights, the diagnostic search for ALS has expanded to include biomarkers such as total tau (tTau) and the phosphorylated tau:tTau ratio in CSF, which can serve as diagnostic markers of ALS. The CSF level of tTau at diagnosis may also play a relevant prognostic role in this disease.75 Furthermore, the presence of TDP-43 in CSF is another useful diagnostic biomarker, particularly given its predominant aggregation in ALS cases. The combined use of CSF NfL and CSF TDP-43 may further enhance the biomarker-driven diagnosis of ALS, integrating these molecular disruptions into a clinical framework.78 Additionally, recent findings from the Pre-fALS study underscore the potential of NfL as a significant biomarker for early ALS detection, particularly in genetically predisposed individuals.79 Elevated levels of NfL were observed up to 12 months before the clinical onset of ALS in carriers of specific genetic mutations, such as SOD1 A4V, indicating its utility in identifying early neurodegenerative changes prior to symptomatic ALS.80 This highlights NfL’s potential as an early intervention tool that could be crucial in modifying disease progression, especially when used in conjunction with other CSF biomarkers, such as tTau and TDP-43.
RNA metabolism
Both FUS and TDP-43 are RNA-binding proteins. This means that they bind to RNA molecules and participate in various stages of their metabolism, covering all steps related to RNA synthesis, modification, processing and regulation. This includes the transcription of DNA into RNA, RNA splicing to form mRNA, which can be translated into proteins, and the transport of RNA within cells.81
In patients with ALS, nucleotide sequence variants in the genes encoding FUS and TDP-43 can lead to the production of proteins that are improperly compartmentalized within the cell. FUS and TDP-43 are normally located predominantly in the cell nucleus, where they play important roles in RNA processing. However, variants can lead to abnormal accumulation of these proteins in the cytoplasm, resulting in subsequent complications that negatively affect RNA-processing mechanisms.71 This ultimately results in the synthesis of defective proteins, contributing to the degeneration of motor neurons.82,83
To summarize the disruptions in RNA metabolism regulation in the pathogenesis of ALS, the following changes can be identified.
-
Transcription defects: issues with the process of copying DNA into RNA.84
-
Splicing changes: errors in connecting RNA segments that affect protein synthesis.85
-
MicroRNA biogenesis: disruption of the creation of microRNAs, which regulate gene expression.86
-
Formation of stress granules: abnormal accumulation of proteins and RNA in stress granules, which are involved in the stress response of the cell.87
-
Nucleocytoplasmic RNA transport: disruption of the transport of RNA between the nucleus and cytoplasm, impairing normal cellular function.87
In light of these disruptions, noncoding RNAs, specifically microRNAs, exhibit considerable promise as biomarkers for ALS. Advanced RNA-sequencing techniques have facilitated the identification of microRNAs that are differentially expressed in the CSF and blood of patients with ALS.88,89 These findings underscore the ability of microRNAs to mirror RNA processing irregularities, for example, those caused by pathologies associated with FUS and TDP-43 proteins. Further investigative efforts are imperative to delineate consistent diagnostic profiles on the basis of miRNA expression levels.
Excitotoxicity
Glutamate activates N-methyl-D-aspartate (NMDA) and AMPA receptors, which mediate the influx of Ca2+ and Na+ into postsynaptic neurons. Excess glutamate leads to abnormal activation of these receptors, causing excessive Ca2+ influx, which results in excitotoxicity, potentially linked to a number of pathological conditions, including ALS.90 Notably, glutamate-regulated AMPA receptors are quite prevalent in motor neurons.91 Experimentally, excessive activation of AMPA receptors leads to hindlimb paralysis and degeneration of motor neurons in wild-type rats, highlighting the susceptibility of motor neurons to disturbances in Ca2+ influx.92 The regulation of Ca2+ permeability by AMPA receptors is mediated by the presence of the GluA2 subunit, whose reduced expression leads to increased permeability of this ion.93
Notably, the GluA2 subunit, when correctly transcriptionally edited, makes the AMPA receptor impermeable to Ca2+. This means that receptors containing a transcriptionally edited GluA2 subunit do not allow Ca2+ to penetrate postsynaptic neurons, preventing the potentially toxic effects of excessive influx of this ion.94 The transcriptional editing process itself involves altering the RNA sequence encoding GluA2, specifically at the position determining Ca2+ permeability. This change is mediated by an enzyme called adenosine deaminase acting on RNA 2 (ADAR2).95 In patients with sALS, there is a reduction in ADAR2 expression.96 This means that less ADAR2 is available for the transcriptional editing of the GluA2 subunit. As a result, a greater number of AMPA receptors may become permeable to Ca2+, which can ultimately lead to excitotoxicity. It has also been shown that reduced levels of transcriptional editing of GluA2 due to decreased ADAR2 expression lead to increased aggregation of TDP-43 in spinal motor neurons.70 The link between reduced ADAR2, improper transcriptional editing of GluA2 and aggregation of TDP-43 points to a complex molecular pathway that contributes to the development of ALS through excitotoxicity.
Thus, reducing excitotoxicity, aimed at decreasing motor neuron damage, underlies the mechanism of action of riluzole. This drug, which became the first medication approved by the FDA for the treatment of ALS in December 1995, works by suppressing glutamatergic neurotransmission. It stabilizes voltage-dependent sodium channels in their inactive state and affects guanine nucleotide-binding processes, which leads to the inhibition of glutamic acid release and the blockade of changes in the NMDA receptors of the postsynaptic membrane.97 Notably, the clinical efficacy of riluzole has been highlighted by a population study that provides real-world data comparing patients treated with riluzole to those not receiving the drug. This study revealed that patients with ALS treated with riluzole exhibit a median survival benefit ranging from 6 to 19 months, which significantly exceeds the 2–3-month benefit initially reported in pivotal randomized controlled trials.98
Oxidative stress
Oxidative stress is an important factor in the initiation of ALS pathogenesis, arising from a disruption in the balance between the formation and neutralization of reactive oxygen species (ROS).99 Interest in this factor first emerged following the discovery of nucleotide sequence variants in SOD1 in patients with fALS.32 Notably, elevated levels of oxidized forms of proteins, RNA, DNA and lipids were recorded in the postmortem tissue from both patients with sALS and those linked to the SOD1 variant, underscoring the critical role of oxidative stress in the development of the disease.100
SOD1 is a key antioxidant enzyme that is widely distributed in the body and catalyses the conversion of superoxide anions into molecular oxygen and hydrogen peroxide.101 In studies conducted among patients with fALS associated with nucleotide sequence variants in SOD1, a significant reduction of 42% in the overall activity of the SOD1 enzyme was observed, which can lead to an imbalance between the production and decomposition of ROS.74 This situation may be further exacerbated by disruptions in the nuclear erythroid 2-related factor–antioxidant response element pathway, which plays a key role in regulating the production of proteins that protect against oxidative stress in SOD1-related ALS.73
In May 2017, the FDA approved the drug edaravone for the treatment of ALS.102 Importantly, the European Medicines Agency (EMA) did not authorize the pharmaceutical under discussion, specifically requesting an additional 1-year placebo-controlled clinical trial to evaluate survival outcomes. Consequently, in 2019, Mitsubishi Tanabe Pharma Corporation chose to retract its marketing authorization application (MAA) for edaravone as an ALS treatment from consideration by the EMA.103 Notably, data from a multicentre cohort study, which used real-world clinical data, compared the long-term safety and efficacy of combined intravenous edaravone and riluzole therapy with those of propensity score-matched controls who received only riluzole therapy among patients with ALS. Although the prolonged administration of intravenous edaravone in patients with ALS has been proven to be feasible and predominantly well tolerated, it has not demonstrated any disease-modifying advantages. Therefore, intravenous edaravone may not offer a clinically significant additional benefit over standard monotherapy with riluzole.104
This medication helps protect neurons in the brain and spinal cord by neutralizing ROS, such as hydroxyl and peroxyl radicals, hydrogen peroxide and peroxynitrite, which contribute to the progression of neurodegeneration.105 Additionally, its protective effect is partly due to the activation of the Nrf2/HO-1 pathway, which reduces cognitive impairments and protects cells from apoptosis.106
NADPH oxidase (NOX) catalyses the formation of ROS, especially those generated by the phagocytic isoform NOX2. Inactivation of NOX2 in transgenic SOD1 mice with ALS reduces ROS production and prolongs survival.107 In studies in a human model, the activity of NOX2 was not dependent on sex, age, duration of the disease, phenotype or data from the ALS Functional Rating Scale-Revised (ALSFRS-R). However, patients whose NOX2 activity was below the median value experienced an increase in survival by 1 year from the onset of the disease.108 These results are consistent with observations in a mouse model of ALS and demonstrate the potential role of NOX2 in the progression of the disease in patients with ALS.
Interestingly, the protein ataxin-2 (the result of ATXN2 expression), which contains a polyglutamine (PolyQ) sequence that is beyond the normal range of length due to expansion (an increase in the number of glutamines), specifically between 27 and 33 glutamine residues (27–33Q), has been identified as a significant risk factor for the development of ALS.109,110 Medium-length PolyQ expansions in the ataxin-2 protein may interact with NADPH oxidase, increasing the activity of this enzyme and leading to increased production of ROS, DNA damage and mitochondrial distress.111
Nucleotide sequence variants in genes associated with ALS, such as NEK1, C21ORF2 and SETX, increase the likelihood of developing ALS due to disrupted regulation of DNA damage repair mechanisms. This, in turn, leads to impaired ability of motor neurons to cope with oxidative stress and, consequently, to cell death.112–114
Conclusion
The aetiopathogenesis of ALS remains complex and not fully understood, despite advancements in genetic research and molecular biology. Key genetic variants in nucleotide sequences in genes such as SOD1, C9ORF72, TARDBP and FUS significantly contribute to disease mechanisms by disrupting RNA metabolism and protein homeostasis, contributing to defects in nucleocytoplasmic transport, impairing DNA repair and participating in excitotoxicity and oxidative stress, which ultimately leads to motor neuron degeneration and highlights the multifaceted nature of ALS pathogenesis. Moreover, the same variants can impact multiple cellular pathways, underscoring the interconnected and complex nature of pathogenic processes in ALS.
The data clearly demonstrate a close connection between the impact of various environmental factors and the increased risk of developing ALS, highlighting the acute need for a better understanding of risk factors associated with the environment. This understanding is important not only for the prevention of this neurological disease but also for developing effective strategies to reduce risks, especially in the most vulnerable population groups. Moreover, the interaction between environmental factors and genetic structure through epigenetic mechanisms further underscores the complex nature of ALS. Thus, ongoing research on the environmental impact on ALS development, combined with in-depth studies of epigenetic changes, plays a key role in understanding the disease’s mechanisms.
Given the convergence of these multiple pathogenetic pathways, it may be prudent to develop biomarkers that reflect these complex interactions. These biomarkers could then be used in combination drug trials aimed at targeting multiple pathways simultaneously. Such an approach could increase the precision and efficacy of treatment strategies, increasing the quality of available treatment options for ALS. Importantly, a clear understanding of these complex molecular and epigenetic interactions and pathways not only enhances our grasp of potential therapeutic targets but also may improve the diagnostic and treatment processes for ALS. Research into the environmental impacts and epigenetic changes associated with ALS is crucial for unravelling these complex mechanisms and for the successful implementation of these advanced therapeutic strategies.