The coronavirus disease 2019 (COVID-19) pandemic has led to unprecedented illness and death among the global population. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection can range from mild upper respiratory symptoms that subside with no lasting effects to multiorgan system failure and death.1 It can also result in numerous neurological symptoms that are less well understood.2 The complete mechanism of these manifestations has yet to be elucidated, although significant progress has been made.3 The complement system is part of natural human immunity and consists of a series of more than 20 proteins, which progress through proteolytic pathways for host defence and regulation.4 This system has been implicated in the pathogenesis of SARS-CoV-2 infection through clinical studies and observations of elevated complement levels.5 This article broadly overviews the current state of knowledge on the neurological manifestations of SARS-CoV-2 infection. We discuss the current understanding of the pathogenesis of complement overactivation in COVID-19 and describe how each consequence may be related to these neurological manifestations. We also review current therapeutic avenues in this regard.
Neurological manifestations of SARS-CoV-2 infection
The neurological symptoms of SARS-CoV-2 infection can be broadly divided into central nervous system (CNS) and peripheral nervous system (PNS) complications. CNS complications include encephalopathy and encephalitis, cerebrovascular disease and headaches, and other, rarer, complications.3,6–8 PNS complications include neuromuscular disease, autonomic dysfunction, and altered taste and smell sensations. Psychiatric symptoms such as depression, psychotic symptoms, anxiety, hallucinations and altered mood are also associated with SARS-CoV-2 infection;6 however, for the purpose of this review, discussion of these is deferred.
Central nervous system involvement in SARS-CoV-2 infection
Non-vascular central nervous system complications
Symptoms of encephalopathy and encephalitis due to SARS-CoV-2 infection include confusion, lethargy, delirium, somnolence and coma. These symptoms in patients with SARS-CoV-2 infection usually occur due to systemic illness resulting in a metabolic encephalopathy, most commonly from hypoxia or renal dysfunction.9 The risk factors are in line with established risk factors for encephalopathy, which include old age, dementia and a history of cerebrovascular disease.9 The overall pooled prevalence of these symptoms of encephalopathy and encephalitis in patients with COVID-19 as a whole has been estimated to be between 7–11%.8,10 The severity of SARS-CoV-2 infection has also been associated with an increased risk of developing these symptoms. Patients who are critically ill with COVID-19 have a much higher incidence of delirium and coma, with sedation due to mechanical ventilation playing a significant role.7,10 Headaches are associated with SARS-CoV-2 infection and are postulated to be related to fevers, systemic inflammatory mediator release or direct viral invasion of nerve endings.11 Headaches occur in 11–14% of patients with SARS-CoV-2 infection and are gradual in onset, moderate or severe, and bilateral headaches around the forehead, temporoparietal and periorbital regions.12
Additionally, acute necrotizing encephalopathy, leukoencephalopathy and demyelinating lesions in the brain and spine, with multiple focal lesions and necrosis in various locations, which are likely to be related to cytokine storm, have been reported after SARS-CoV-2 infection.13
Vascular central nervous system complications
Cerebrovascular disease due to SARS-CoV-2 infection comprises primarily ischaemic and haemorrhagic strokes and cerebral venous thrombosis.14 The cause of ischaemic stroke is currently unclear and likely multifactorial, but hypotheses include a prothrombotic and hypercoagulable state due to inflammatory cytokine storm, viral invasion resulting in endothelial dysfunction and vasoconstriction, and hypoxaemia leading to the activation of hypoxia genes and thrombotic events.15 Haemorrhagic stroke, which is less common, is hypothesized to be related to angiotensin-converting enzyme-2 (ACE2) receptors in blood vessel walls, which allow the virus to damage and rupture vessels, along with the downregulation of the renin-angiotensin system, which increases blood pressure; the viral infection may also be related to the consumption of coagulation factors.14 Cerebral venous thrombosis is associated with SARS-CoV-2 infection through the classic mechanism of hypercoagulable state, along with endothelial damage and stasis leading to the generation of thrombi.16 These haemostatic abnormalities are demonstrated by increased fibrin degradation products and D-dimers, as well as intravascular coagulopathy.17 In patients with SARS-CoV-2 infection, the pooled prevalence is 1.00% for ischaemic stroke, 0.31% for haemorrhagic stroke and 0.12% for venous thrombosis.10 Ischaemic stroke has been linked to hypercoagulability, vasculitis and cardiac dysfunction.18 Haemorrhagic complications are possibly due to endothelial rupture from viral neuroinvasion, secondary haemorrhagic transformation of ischaemic strokes or massive cytokine release and breakdown of the blood–brain barrier.18
Peripheral nervous system involvement in SARS-CoV-2 infection
Muscular complications
Muscle damage such as rhabdomyolysis and myalgias, which can be caused by generalized inflammation, may be associated with SARS-CoV-2 infection through a form of immune response known as cytokine storm.6 The pooled prevalence of muscle damage among all patients with COVID-19 is reported to be 20.00% for myalgia, 5.00% for skeletal muscle injury and 0.39% for rhabdomyolysis.6,10 Direct viral invasion of the muscle has also been hypothesized, leading to myositis and breakdown.19 Patients who are critically ill with COVID-19 and require mechanical ventilation with specific neuromuscular blocking agents have been observed with myopathy; however, this is not specific to COVID-19.6
Nerve-related complications
PNS nerve complications associated with SARS-CoV-2 infection include inflammatory neuropathies such as Guillain–Barré syndrome (GBS) and Miller Fisher syndrome (MFS), autonomic neuropathy, small fibre neuropathies and other neuropathies causing neuropathic pain, and cranial neuropathies causing anosmia and ageusia.6 Molecular mimicry between viral proteins and peripheral nerve proteins resulting in damage to neuronal myelin and axons has been found to be the established mechanism of GBS and MFS after any viral illness, including SARS-CoV-2 infection.6 GBS after SARS-CoV-2 infection has been described with lower-limb weakness and, eventually, generalized weakness and paralysis of all four limbs, and MFS is recognized as the variant with ophthalmoplegia, areflexia and ataxia.2 The pooled prevalence among all patients with COVID-19 is 0.28% for GBS and 0.05% for MFS.10
Anosmia and ageusia are well-known symptoms of SARS-CoV-2 infection.20 The mechanism for these effects is hypothesized to be a direct viral invasion of the olfactory epithelium due to ACE2 neurotropism.21 In patients with COVID-19, impairment of smell has a pooled prevalence of 19%, and loss of taste has a pooled prevalence of 21%,10 with an earlier study estimating the combined loss of taste and smell to have a pooled prevalence of 35%.22 Anosmia has been associated with milder cases and is a prognostic factor for lower mortality due to SARS-CoV-2 infection.23
Autonomic dysfunction includes changes in blood pressure, heart rate, respiratory rate and temperature and can manifest with symptoms including fatigue, bladder dysfunction and gastrointestinal symptoms such as nausea.24 These symptoms are reported during both the acute phase of SARS-CoV-2 infection and in the weeks to months after the viral illness has passed.25 The mechanism of these symptoms is postulated to be related to viral invasion and inflammation of the median eminence of the hypothalamus and the brainstem resulting in the release of hormonal mediators. It could also be related to autoantibodies against certain receptors.25
Rarer neurological manifestations of SARS-CoV-2 infection
Rarer neurological complications of SARS-CoV-2 infection have been reported, including encephalitis and meningitis.7 The description of these cases varies, ranging from typical to atypical; there are also disparate reports about the detection of SARS-CoV-2 in the cerebrospinal fluid of patients with these diseases.7 An autoimmune cause of encephalitis as a post-infectious syndrome has been suggested.26 Seizures have been reported as well, but these could be related to febrile illness, as they are very infrequently reported in COVID-19 cohorts. For example, in a study of 6,147 patients with COVID-19, the rate of seizures was only 0.08%.27
Pathogenesis of neurological manifestations of SARS-CoV-2 infection: excessive complement activation
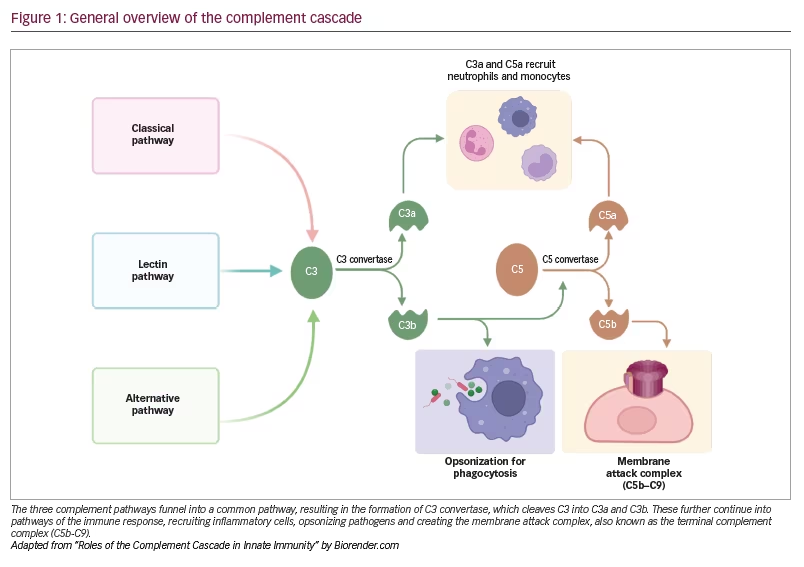
The complement system is an essential and critical component of the innate and adaptive immune response in humans that works via opsonization, inflammatory mediation and cytolysis. This system comprises a series of proteolytic reactions that is activated through three pathways: classical, lectin and alternative.4 These different pathways funnel into a common pathway, resulting in the formation of C3 convertase, which cleaves the C3 protein leaving the C3a and C3b components.4 C3a is a proinflammatory molecule, and C3b plays a role in opsonizing pathogens and forming C5 convertase, which releases C5a, a proinflammatory molecule, and C5b, which forms part of the membrane attack complex (C5b-C9) (Figure 1).4
Although this system is beneficial and essential in combatting bacterial and viral infections, excessive activation, for example, during SARS-CoV-2 infection, can result in direct host damage. The activation of complement as part of the coronavirus disease process was described in a 2018 study on the earlier SARS-CoV-1, where mice that were C3 deficient demonstrated a milder form of the disease, with lower levels of inflammatory cytokines compared with wild-type mice, even in the presence of equivalent viral loads.28
Evidence for this in clinical studies of SARS-CoV-2 infections implicates complement in the development of COVID-19, as well as in the disease severity. In an ultra-high throughput clinical proteomics study of sera of patients with severe SARS-CoV-2 infection, consistent activation of components of the classical and alternative pathways was observed.29 In a study on five individuals with severe SARS-CoV-2 infection, complement components such as C5b-C9, C4d and mannose-binding lectin-associated serine protease (MASP)-2 were deposited in pulmonary and cutaneous tissue, suggesting that widespread complement activation through the alternative and lectin pathways may cause generalized thrombotic microvascular injury.30 In a prospective single-centre study of 197 patients, C3a, C3c and C5b-C9 were elevated in the plasma of patients with COVID-19 compared with healthy controls, with significantly higher C3a and C5b-C9 concentration in COVID-19 cases in the intensive care unit compared with non-intensive care unit cases and higher mortality in patients with consistently elevated C3a.31 Similarly, C3 hyperactivation was an independent risk factor of mortality in a prospective cohort study of 102 inpatients with COVID-19.32 In a prospective cohort study of 39 patients with SARS-CoV-2 infection, C5b-C9 and C4d at admission were significantly elevated in patients with respiratory failure compared with patients without respiratory failure.33 These elevated levels were found days to weeks after admission, demonstrating persistent activation. In a study of 72 patients with COVID-19, a genetic variant at a specific multigene cluster was associated with increased SARS-CoV-2 infection severity and increased activation of complement, with high circulating C5a and C5b-C9.34 Elevated C5a was used in a prognostic model for SARS-CoV-2 infection that predicted severity and mortality significantly.35
Overall, C5a, C3a and C5b-C9 clearly increase in SARS-CoV-2 infection and with disease severity in the literature.36 Even compared with other viruses, such as influenza leading to acute respiratory failure, complement activation is higher and distinguishes patients with worse outcomes.5 However, it has been hypothesized that this aberrant dysregulation of complement may simply lead to greater susceptibility to severe disease.36 Nevertheless, excess activation of complement is clearly involved in severe COVID-19, with the precise mechanism still under investigation.
Mechanism of activation of complement in SARS-CoV-2 infection
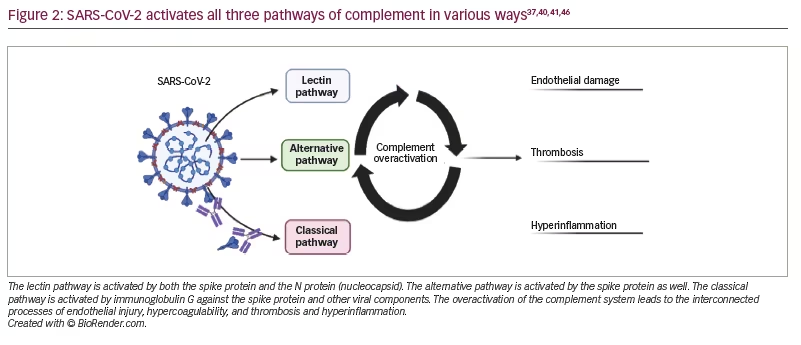
The mechanism of complement activation through the lectin pathway in SARS-CoV-2 is related to lectin pathway recognition molecules ficolin-2, collectin-11 and MASP-2 binding to the S and N proteins of SARS-CoV-2.37,38 Subsequently, through the lectin pathway, the deposition of C3b and C4a occurs.37 Further, the SARS-CoV-2 S protein, but not the N protein, activates the alternative pathway by interacting with the cell surface and binding to heparan sulphate, allowing for the generation of C3 convertase.39 Moreover, the SARS-CoV-2-induced expression of C3 through a Janus kinase signal/transducer and activator of transcription pathway leads to activating C3a through the alternative pathway component complement factor B.40 Specifically, components of the alternative pathway are associated with worse outcomes for patients with SARS-CoV-2 infection and are associated with markers of endothelial damage and hypercoagulability.5 The classical pathway may also be activated through a direct viral invasion of endothelial cells, through ACE2 receptors by SARS-CoV-2, and through the exposure of damage-associated molecular patterns.41 Immunoglobulin G against SARS-CoV-2 activates the classical pathway, resulting in excess inflammation.42 Mannose-binding lectin has been suggested to play a role in thrombosis in SARS-CoV-2 infection; once bound to the target, it circulates with MASPs, which activate complement and are also involved in clotting.43 C5a, with its receptor C5aR and C3a, have been postulated in SARS-CoV-2 infection to increase the recruitment of myeloid cells,44 which then secrete inflammatory molecules such as interleukin 6, tumour necrosis factor a, and interleukin 1b, leading to a cytokine storm.45 In general, this activation may further lead to endothelial damage, inflammation and thrombosis, all of which may be implicated in the various neurological manifestations of SARS-CoV-2 infection (Figure 2).37,40,41,46 We review each consequence of complement overactivation in the context of the neurological manifestations of SARS-CoV-2 infection.
Endothelial damage in the central nervous system
Complement overactivation results in endothelial injury and reveals the basement membrane, which can lead to the activation of clotting factors.46 In a study using a mouse model, complement activation was found to contribute to endothelial dysfunction, and SARS-CoV-2 infection was found to upregulate the kirsten rat sarcoma pathway.47 This pathway increases the expression of serpin genes, which were similarly upregulated in the blood–brain barrier of mouse multiple sclerosis models, indicating a loss of the endothelial barrier function and vascular permeability.47 Disruption of the vasculature in the brain through an unknown mechanism has been described in postmortem studies of the brains of patients with COVID-19.48 A current theory for ischaemic and haemorrhagic strokes in SARS-CoV-2 infection is the direct viral invasion through disruption of the blood–brain barrier.49,50 A hypothesized pathway involves the zonulin protein, a regulatory protein of tight junctions in various tissues that is known to induce complement, the overactivation of which has been linked to increased permeability and, possibly, to SARS-CoV-2 neuroinvasion.51 Neuroinvasion is thought to result in cerebral sinus venous thrombosis, as well as the meningitis and encephalitis seen in SARS-CoV-2 infection.52 Additionally, complement-mediated endothelial damage through C5b-C9 has been linked to the release of extracellular vesicles, which increase the prothrombotic environment.53 These vesicles could serve as markers of endothelial injury and ischaemic stroke in SARS-CoV-2 infection.53 In a recent autopsy study of the brains of nine patients with COVID-19 by Lee et al., disruption of the blood–brain barrier, implied by the presence of large proteins, which do not usually cross the barrier, was found in all patients.54 This endothelial dysfunction was suggested to be complement mediated, as the deposition of complement components was observed. Lee et al. proposed that SARS-CoV-2 infection triggers immune complexes through the classical pathway, which cause multiple areas of endothelial activation leading to platelet activation, thrombi and, eventually, inflammation; this results in microglia activation, which causes neuronal injury.54 A disordered activation of complement, which can occur due to SARS-CoV-2, can trigger neuroinflammation through nuclear factor k-light-chain-enhancer of activated B cells, microRNA and caspases, leading to apoptosis in the brain.55
Hyperinflammation in the central and peripheral nervous systems and muscle
The elevated levels of proinflammatory cytokines in patients with COVID-19 and, specifically, more severe types may be promoted by C3a and C5a, as previously mentioned. However, this is likely only a component of the process, as a whole host of cytokines work together in a cascade, leading to massive multiorgan inflammation.45 This cytokine storm is associated with the autonomic dysfunction that has been observed in SARS-CoV-2 infection through increased sympathetic activation and parasympathetic withdrawal.56 Additionally, complement activates neutrophil extracellular traps (NETs), which can lead to autoimmunity, manifestations of which include GBS, myositis and myalgia,57 although this link has yet to be investigated. C3a, C5a and C5b-C9 are all elevated in the cerebrospinal fluid of patients with GBS and have a role in axon damage and demyelination.58 Pathological specimens of nerves from patients with GBS have deposits of C3 and C5b-C9, which have been suggested to be involved in the damage.59 This deposition leads to myelin damage and infiltration with inflammatory cells.59 Moreover, anosmia and ageusia associated with COVID-19 are thought to be due to the viral invasion of the neuro-epithelium.60 Similarly, smell dysfunctions have been reported in autoimmune diseases characterized by the overactivation of the complement system, suggesting that this excess complement activation could play a role in anosmia.60 Despite the suggestion that pulmonary infection is the main cause of death due to COVID-19, cytokine storm has been suggested as a mechanism of pathological changes in the brain.47 Interestingly, complement has been hypothesized to be related to the delirium observed in patients who are critically ill with COVID-19 through hyperinflammation.61
Complement has been clearly described in the mechanism of inflammatory myopathies and has been suggested to be involved in SARS-CoV-2 infection as a trigger of these myopathies.62 C5b-C9 is deposited on endothelial cells in patients with dermatomyositis even before muscle destruction has occurred; the activation of complement recruits inflammatory cells leading to endothelial cell necrosis and microinfarcts.59 A similar mechanism may be present in patients with COVID-19, leading to the wide spectrum of muscle damage observed.
Hypercoagulability and thrombosis in the central nervous system
Currently, a ‘two-hit’ hypothesis has been proposed to describe the link between complement and thrombosis in COVID-19, wherein the first relates to mutations or acquired changes in complement regulatory proteins or C3 convertase, and the second is an inflammatory trigger.63 This link in SARS-CoV-2 infection is commonly compared to thrombotic thrombocytopenic purpura, where a genetic disposition and subsequent inciting factor lead to thrombosis.64 Although it is still unknown why some patients develop mild versus severe SARS-CoV-2 infection, a possible explanation could be variations in complement genes leading to stronger complement activation, excess complement deposition and progressive thrombotic microangiopathy (TMA), and, thus, worse outcomes.65 TMA leads to decreased blood flow, resulting in ischaemia, injury and eventual organ failure. Indeed, single-nucleotide polymorphisms of complement regulators, which allow for overactivation, are associated with adverse outcomes in SARS-CoV-2 infection.66 Diffuse microvascular thromboses have been found in the autopsy of patients with SARS-CoV-2 infection and are similar to those found in SARS-CoV-1 infection, including those found in the brain.67
Complement shows considerable interaction with coagulation pathways, and both are closely linked with significant crosstalk, such that it has been suggested that both pathways are evolutionary descendants of a common immune proteolytic system.68,69 The interactions between complement and the clotting and coagulation pathways include thrombin activating C5 independently of C3 and high levels of von Willebrand factor (vWF), factor XII, kallikrein, and platelets, which trigger complement activation and the subsequent release of C5a, C5b-C9 and MASPs, which facilitate thrombosis.64 These pathways lead to an escalating cycle of damage to surrounding tissue and may play a role in the development of large-vessel stroke seen in SARS-CoV-2 infection.64 Furthermore, NETs activated by complement are also implicated in thrombosis in SARS-CoV-2 infection through tissue factor,70 fibrinogen, vWF and fibronectin.71 NETs are associated with more severe SARS-CoV-2 infection and play a role in the occlusion of arteries and venous thrombosis,72,73 and, thus, possibly stroke.68,74 In a case report of a patient with COVID-19 and complement-mediated thrombosis, it was hypothesized that the transient elevation of anti-phospholipid antibodies triggered by SARS-CoV-2 may have contributed to coagulopathy.75 As complement activation is necessary for anti-phospholipid-mediated thrombosis,75 this could serve as another mechanism of stroke. The kallikrein-kinin system (KKS) is another part of the immune system and has extensive crosstalk with complement in a manner that has led to hypothesizing that KKS is overactivated in SARS-CoV-2 infection, exacerbating hypercoagulation.76 KKS is an important driver of thrombus formation in stroke.77 Clinically, coagulopathy in patients with COVID-19 is usually characterized by elevated D-dimer, fibrinogen, vWF and factor VIII, and only mildly elevated or normal platelet count, activated partial thromboplastin time and prothrombin time, suggesting a pattern similar to that established in complement-associated TMA.64,78
Excess complement after coronavirus disease 2019 vaccination leading to neurological complications
COVID-19 vaccination is associated with the development of stroke, specifically ischaemic stroke and cerebral venous thrombosis, although the overall reporting rate of stroke did not increase during the vaccine rollout.79,80 It has been hypothesized that the immune response to the vaccine can overactivate complement, leading to endothelial disruption and hypercoagulability and creating the necessary environment for stroke.81 Additionally, in a case of COVID-19 vaccine-induced thrombotic thrombocytopenia, compared with controls, complement components were detectable in the vaccine-induced thrombotic thrombocytopenia thrombi, implicating complement in the pathogenesis.82 The hypothesized mechanism through which the COVID-19 vaccine overactivates complement is related to anti-platelet factor 4 autoantibodies, which could facilitate the activation of C3. Additionally, the binding of complement to the anti-platelet factor 4 immune complexes can lead to complement activation through the classical pathway.83 A variety of neurological adverse events are associated with COVID-19 vaccination; these events include stroke, encephalopathy, GBS and rhabdomyolysis.84 Complement may play a role, though further investigation is needed.
Excess complement in neurological post-acute sequelae of coronavirus disease 2019
Long-haul COVID-19, also known as long COVID and post-acute sequelae of COVID-19, is a collection of heterogeneous symptoms and complications that follow the acute infectious phase, usually 4 weeks, and that last for more than 6 months.85 Complement elevation with sustained inflammation has been observed in patients with long-haul COVID-19. Long-term respiratory symptoms, for example, are associated with high C5a levels, suggesting a mechanistic function as well as a potential biomarker.86 Additionally, elevated C3 has been seen in patients with persistent lung abnormalities after COVID-19.87 Neurological symptoms of long-haul COVID-19 include CNS symptoms such as ‘brain fog’, difficulty concentrating, memory problems, anxiety, and insomnia and PNS symptoms such as autonomic dysfunctions, neuropathic pain, muscle damage, and peripheral neuropathies.88 Complement overactivation and deposition in the brain, which lead to inflammation, damage and neuron death, have been postulated by Lee et al. to be involved in the progression of long-haul COVID-19.54 In their study, the authors found pathological findings from SARS-CoV-2 infection, particularly in the hindbrain, that could have come from the viral invasion of the brainstem through the vagus nerve.54 The vagus nerve is involved in both respiratory and gastrointestinal function, and viral invasion and subsequent hyperinflammation could explain the persistent autonomic dysfunction observed in long-haul COVID-19.54,56
Complement inhibition: a potential therapeutic option for SARS-CoV-2 infection
The current landscape of complement inhibition as an intervention for SARS-CoV-2 infection primarily uses existing drugs for complement-mediated diseases. These diseases include complementopathies such as macular degeneration, atypical haemolytic uraemic syndrome, paroxysmal nocturnal haemoglobinuria, complement nephropathies and myasthenia gravis.89 Inhibitors of C5a, such as eculizumab, have shown varying but positive results. Eculizumab treatment improved survival in 35 patients,90 decreased mortality and discharge with complications in 10 patients,91 decreased inflammatory markers in four patients,92 improved clinical status in eight pregnant patients and improved symptoms in 13 patients.93,94 However, in a clinical trial of 135 patients, another C5 inhibitor, ravulizumab, did not show clinical benefit, and the trial was terminated.89 C5 inhibition has and is being studied with eculizumab (Eculizumab [Soliris] in COVID-19 infected patients [SOLID-C19]; ClinicalTrials.gov identifier: NCT04288713; CORIMUNO19-ECU: Trial evaluating efficacy and safety of eculizumab [Soliris] in patients with COVID-19 infection, nested in the CORIMUNO-19 cohort; ClinicalTrials.gov identifier: NCT04346797), tesidolumab95 and peptide antagonists such as nomacopan96 and zilucoplan (Zilucoplan® in improving oxygenation and short- and long-term outcome of COVID-19 patients with acute hypoxic respiratory failure [ZILU-COV]; ClinicalTrials.gov identifier: NCT04382755).89
C3 inhibition using AMY-101 has been reported in four patients with SARS-CoV-2 infection, all of whom recovered.97,98 However, a phase I/II clinical trial (A study of APL-9 in adults with mild to moderate ARDS due to COVID-19; ClinicalTrials.gov identifier: NCT04402060) in 65 patients treated with the C3 inhibitor APL-9 found no difference in mortality.89 In six patients, inhibition of MASP-2, which is involved in the lectin pathway, using narsoplimab resulted in the survival of all patients with severe SARS-CoV-2 infection.99 Conestat alfa, a C1 inhibitor from the classical pathway, was used in five patients and resulted in clinical improvement and recovery in four patients.100 However, these positive results must be weighed against an increased risk of severe bacterial infections. Several patients on eculizumab developed bacterial infections, in some cases resulting in death.36 These infections occurred even in the presence of antibiotic prophylaxis and vaccination.89
Conclusions
The aberrant dysregulation of the complement system resulting in excess activation clearly plays a role in at least a subset of patients with SARS-CoV-2 infection. Genetic susceptibilities of variants and regulators of complement genes, as well as specific risk factors, may determine which patients develop a severe infection. Through complement, the interconnected triad of endothelial damage, thrombosis and hyperinflammation is undoubtedly involved in the development of certain neurological manifestations of SARS-CoV-2 infection, especially cerebrovascular disease. The increased inflammation and cytokine storm associated with complement likely plays a role in manifestations such as muscle and nerve injury, although studies have yet to explore this link in depth in the context of SARS-CoV-2 infection. Neurological manifestations result in enormous morbidity for patients with COVID-19, and there are still limited data on their long-term effects; therapies targeting complement may mitigate these effects. Currently, however, complement inhibition requires more randomized clinical trials before being recommended to all patients with COVID-19, if at all. It is likely that once more is elucidated on the mechanism and involved pathways, complement inhibition may be the choice for selected patients with COVID-19.