Definitions and Epidemiology
An accurate diagnosis is necessary before appropriate treatment can be initiated. In contrast to most neurological entities, there is no universally accepted definition of status epilepticus (SE). Broadly speaking, SE is the occurrence of continuous seizures or of two or more seizures without intercurrent restoration of neurological function. While epidemiological studies use a timeframe of 30 minutes, derived from experimental evidence of neuronal damage occurring after 30–60 minutes 1 and a seminal consensus paper published in the early 1990s,2 more recently an operational approach defining SE after five minutes has been advocated.3 This encourages prompt medical management, since most convulsive seizures end before two minutes.4 However, it has been shown that up to 40% of seizures lasting between 10 and 30 minutes abort spontaneously,5 tending to favor the more conservative definition. Commonly, SE is defined as refractory if it is resistant to two or more antiepileptic drugs (AEDs) administered at adequate dosage either without a minimum duration or, in some frameworks, persisting for at least one hour.2,6
Although every seizure type can theoretically persist and evolve into SE, a simple 2D SE classification system can be constructed, either generalized or focal to be combined with convulsive or non-convulsive.7 While generalized convulsive SE appears straightforward, generalized non-convulsive SE has distinct varieties with different clinical implications. In comatose patients, usually in intensive care unit (ICU) settings and often after cessation of convulsive SE, it may be termed subtle SE.8 Generalized non-convulsive SE can also be a manifestation of
absence SE in patients with idiopathic generalized epilepsy, in which case the patient is typically encephalopathic rather than comatose. A form of focal convulsive SE characterized by a more or less continuous seizure state affecting a restricted part of the body is also labeled ‘epilepsia partialis continua,’ and focal non-convulsive SE may manifest as prolonged or frequently repeated complex partial seizures.
SE is often the expression of underlying severe disturbances of the central nervous system (CNS), and occurs in 10–41/100,000 persons per year according to population-based studies.9–12 Refractory SE, defined as resistant to two or more treatments, develops in about 10–40% of SE cases.13–16
Diagnosis and Differential Diagnosis
Four main criteria contribute to SE diagnosis: clinical presentation, duration, electroencephalogram (EEG), and response to treatment.7 However, diagnosis is often not straightforward, because clinical features are extremely variable, seizure onset is often difficult to determine, EEG pattern tends to evolve with time,8 and treatment response is not uniform. Imitators of SE are usually characterized by abnormal movements and diminished responsiveness. While classic movement disorders have typical features, post-sedation shivering or posturing in the ICU may sometimes prove hard to differentiate from subtle SE, and so-called diencephalic seizures, also termed paroxysmal sympathetic storm, can include tonic posturing or tremor that may mimic convulsions.17 However, the most important entity that needs to be positively identified is prolonged psychogenic non-epileptic seizures.18 These may be frequently mistaken for SE, leading to potentially dangerous medical overtreatment.19,20
General Outline and Pharmacological Background
There is a general consensus on the need to treat SE as soon as possible in order to prevent potentially deleterious sequelae.3,6,21,22 The pathophysiological mechanisms occurring during an episode of SE have been well described in animal studies.1,21,23–29 Briefly, at the beginning there is an imbalance between inhibitory, mostly gamma-amino butyric acid (GABAA), and excitatory, predominantly glutamate, inputs of neuronal circuits. This serves as a rationale to begin SE treatment with benzodiazepines, which are rapidly acting GABA-ergic agents. GABA resistance develops progressively due to GABA receptor internalization and subunit changes; afterwards, a shift towards self-sustaining glutamate-mediated excitotoxicity occurs, resulting primarily from the activation of N-methyl-D-aspartic acid (NMDA) receptors (blocked by magnesium ions under normal conditions). These changes may explain both refractoriness to benzodiazepines and excitotoxic neuronal damage. During the first stage metabolism is hypercompensatory, with arterial hypertension, hyperglycemia, and cerebral hyperperfusion, whereas in the second stage, following a transitional period occurring in experimental animals after 30–60 minutes, there is progressive deregulation leading to cerebral hypoperfusion, lactic acidosis, and multiorgan failure.
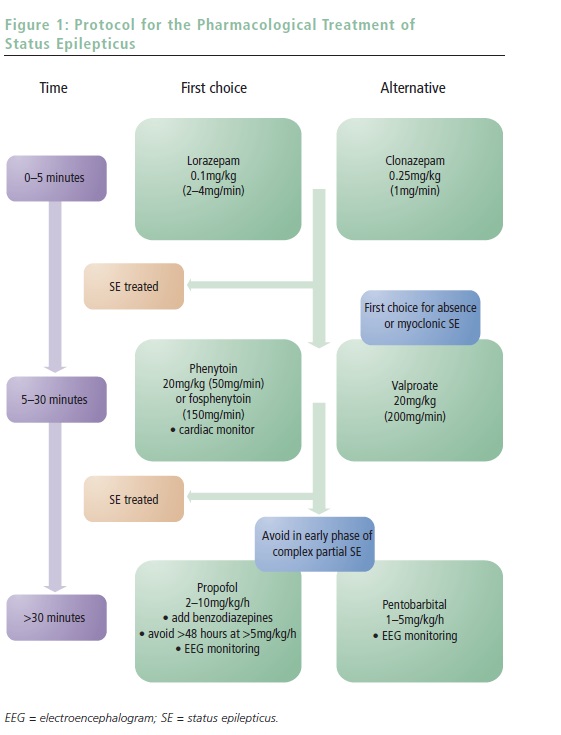
SE treatment may be conceptually categorized into three phases of, usually intravenous (IV) AED administration. The first consists of benzodiazepines and aims at rapid SE control; the second, using classic AEDs, targets early resistant forms and begins long-term coverage following anticipated control of SE; and the third treatment phase is reserved for refractory episodes and comprises administering general anesthetics. A simple protocol with corresponding timing is proposed in Figure 1. Awareness of the protocol greatly facilitates this practical approach and allows smooth interplay between the different providers (paramedics, emergency or ICU team, and neurologists).
First-line Treatment
The first-line treatment of SE has been far better investigated than secondand third-line treatment. A small study in 1983 found a non-significant trend toward better response to lorazepam (LZP) compared with diazepam (DZP).30 A pre-hospital trial found that LZP had a non-significant superiority over DZP, whereas both treatments were significantly better than placebo. Cardiovascular and respiratory complications did not differ among groups.31 A large Veterans Association (VA) trial, focusing on generalized convulsive SE and assessing the efficacy of LZP, phenobarbital (PB), DZP followed by phenytoin (PHT), and PHT alone, found better efficacy of LZP compared with PHT alone, but not compared with the other treatment arms.32 The overall response in overt SE was much higher than in subtle SE (about 60 versus 20%). This study has been criticized for choice of treatment arms (lack of LZP+PHT) and the assessment of efficacy at 20 minutes, before the likely peak action of PHT and PB. Nevertheless, as SE becomes more refractory to treatment with time,7 and the first treatment has a far better chance of success than the second or third, regardless of the drug (55 versus 7 versus 2%),33 it is important to administer IV drugs that act quickly. Therefore, benzodiazepines represent the better option over PB and PHT, although essentially there is usually no contraindication to giving both at the same time. Compounds with a long CNS elimination half-life are desirable, since this avoids rebound seizures as drug levels decline. Note that tonic SE in patients with developmental delay may be aggravated by benzodiazepines.
- LZP is the most commonly used first-line treatment. It is administered in
a slow bolus of 0.1mg/kg (2mg/min), enters the brain in <2–3 minutes,34
and has a long duration of action (approximately 12 hours), as it is far
less prone to redistribute in the tissue than DZP.35 Its elimination half-life
is eight to 25 hours.34,35 - DZP is administered at 0.2mg/kg (5mg/min). It readily enters the brain
(in <10 seconds), but its free fraction promptly redistributes in the fat tissue owing to its high lipophilia and protein binding (99%), so its CNS action after the bolus is limited to about 20 minutes.35 It may also be administered rectally. - Clonazepam (CZP) is neither licensed for this use nor available in IV form
in the US, but is relatively widely used in Europe. It is administered at a
bolus of 0.025mg/kg. It reaches the brain in <1 minute34 and, despite its lipophilia, has stable action over time. It has a long half-life (up to 38 hours) and moderate protein binding (less than LZP: 65 versus 90%).34,36 - Midazolam (MDZ) has a short half-life (about two hours), but represents
a valuable alternative when IV lines are not available, or in children
(intranasal or buccal administration). The usual dosage is 0.1–0.2mg/kg,
but doses up to 0.5mg/kg have been reported.37 - PHT is the most widely used agent in this context, administered at
20mg/kg (maximal infusion rate 50mg/min). It is less lipid-soluble than
benzodiazepines. Considering also the infusion rate, it enters the brain
relatively slowly, and maximal concentrations in the CNS are reached
after 20 minutes.35 The elimination half-life is about 24 hours, and
longer at high levels. Some rare but serious local reactions (purple
glove syndrome) are induced by the alkaline solution, which contains
propylene glycol, whereas PHT itself is associated with a consistent risk
of hypotension and bradyarrhythmia (27 and 7% in the VA study
group).32 Elderly subjects are at increased risk, and a slower infusion
rate may be advisable. Cardiac monitoring should always be available
during intravenous PHT administration. - Phosphenytoin is a water-soluble PHT prodrug that lacks propylene
glycol. Therefore, it is safer in terms of local reactions. It is administered
in PHT equivalents. Although it can be infused at a much faster rate
(150mg PHT equivalents/min), it is questionable whether effective CNS
concentrations are reached before PHT administered at optimal rates.43 - PB is given at 15mg/kg (100mg/min). It is less lipophilic than PHT,35 and
it reaches the brain after 20–40 minutes. Its half-life is long, around 100
hours. It also bears a consistent risk of hypotension (34% in the
VA study).32 - VPA is loaded at 20mg/kg up to 200mg/min,38,44 and its elimination halflife
is about 15 hours, shorter in the presence of hepatic enzyme
inducers. VPA, despite its hydrophilic nature, enters the CNS rapidly
through active transport.45 Clinical experience in SE suggests that
effective CNS concentrations are reached within 30 minutes.40,44 Its main
advantage is the lack of cardio-depressive reactions. - LEV may be loaded up to 20mg/kg 42 and its plasma half-life is about
seven hours, but the bioavailability within the blood–brain barrier is
probably considerably longer.46 It is unclear how fast LEV reaches the
brain, but personal observations suggest that a physiological effect
occurs within 15–30 minutes of IV administration. The most frequent
adverse event is mild sedation, and no cardiovascular adverse
reactions have been reported. - Barbiturates such as thiopental in Europe or its metabolite pentobarbital
(PTB) in North America, show a long elimination half-life—15–22
hours—after continuous administration.58 There is a considerable
tendency of this drug to accumulate, prolonging the need for
mechanical ventilation. Their main action is GABAA-agonistic, with some
modulation of calcium channels,59 and both are NMDA antagonists
in vitro.60 Induction with PTB is performed with boluses of 5–15mg/kg,
and the maintenance dose is 1–5mg/kg/h. - Propofol has a short half-life of about one to two hours,61 allowing rapid
titration and withdrawal. It acts as a GABAA agonist, and modulations of
calcium and sodium channels are also described.62 Its effect on NMDA
glutamate receptors is controversial.60,63 It may induce the so-called
‘propofol infusion syndrome,’ a potentially fatal cardio-circulatory
collapse with lactic acidosis, hypertriglyceridemia, and rhabdomyolysis,
feared especially in young children, which to date has been only
exceptionally described in patients with SE.64,65 The concurrent use of
benzodiazepines seems to lower the needed propofol dose, possibly
reducing the risk of this complication.66 Loading dose is 2mg/kg, or
higher if needed to induce burst-suppression or stop seizures, followed
by maintenance at 2–10mg/kg/h. Prolonged (over 48-hour)
administration of doses >5mg/kg/h should be avoided considering the
risk of propofol-infusion syndrome. - MDZ has an extremely variable half-life after prolonged infusion—six to
40 hours67—with marked tachyphylaxis developing within 24–48 hours.68
Benzodiazepines are GABAA agonists without action on NMDA receptors.
MDZ is loaded at 0.2mg/kg, then maintained at 0.05–0.6mg/kg/h.
The administration of a benzodiazepine bolus may induce respiratory and circulatory collapse (about 10–26%),31,32 thus monitoring of these functions is mandatory. Alternatively, the infusion speed may be slowed and accompanied by gentle fluid expansion, especially in older subjects.
Second-line Treatment
Although several compounds are used at this treatment phase, to date there have not been any large-scale, prospective, comparative assessments among AEDs used as second- or third-line SE treatment. The VA study included a PHT, which acts principally through sodium channel modulation, and a PB (mainly a GABAA agonist) arm as initial SE treatment, and found a non-significant trend toward better efficacy of PB (58 versus 44%).32 Valproate (VPA), a ‘dirty drug’ influencing GABA receptors as well as ion channels, administered intravenously has been increasingly reported to be efficacious for several SE types,38–37 even postanoxic forms,40 without inducing cardiovascular adverse reactions. Unlike PB and PHT, VPA may be given without concurrent cardiac monitoring, even in elderly subjects. Of note, a recent study claiming superiority of VPA over PHT38 was encouraging but methodologically limited.
Levetiracetam (LEV) probably exerts its efficacy through altered neurotransmitter release via binding of a synaptic vesicle protein and has been used in SE treatment.41 The recent availability of an IV formulation makes it an even more promising option.42
Third-line Treatment
Since in generalized convulsive SE the earliest administered treatment has the greatest chance of being effective,28,33 the sequential administration of second-line treatments does not appear to have a good rationale, and it seems reasonable to proceed straight to third-line treatment once a given second-line drug (which takes at least 20–30 minutes to be effective) has failed.6,47 An important exception concerns cases in which mechanical ventilation should be avoided, such as those in which the patient is at least partly conscious, and including nearly all cases of absence and most cases of complex partial SE. Indeed, it is unclear whether prolonged complex partial seizures in humans induce permanent structural neurological damage,48–52 as opposed to generalized convulsive SE, in which damage in the limbic structures has been confirmed both pathologically and radiologically.53,54 Thus, it is debatable whether and when coma induction is warranted in SE forms other than generalized convulsive and subtle SE, as it may predispose to several complications, e.g. pneumonia, deep vein thrombosis, pulmonary embolism, neuropathy, and myopathy. In some situations, VPA or LEV may represent valuable, relatively non-sedating options. If this course is chosen, it is important to treat promptly and at adequate doses. Of note, SE episodes in patients with idiopathic generalized epilepsy (absence or myoclonic SE) readily respond to benzodiazepines and VPA. On the other hand, post-anoxic SE is the expression of a severe underlying encephalopathy and is often refractory to standard treatments. In selected cases it may be advisable, after considering other prognostic factors, to administer a short-acting anesthetic for 24 hours before re-assessing the patient.55 Keeping in mind the pathophysiological considerations, a drug with NMDA antagonist activity should be administered. However, since pure NMDA antagonists may induce neuronal damage, a drug with simultaneous GABA-agonist activity may be desirable.24
To date, no prospective, controlled trial has been conducted at this phase of SE treatment. Existing studies on refractory SE are case series. A metaanalysis of barbiturates, propofol, and midazolam56 did not disclose any significant difference in short-term mortality among these three agents, although discrepancies were noted in both efficacy and tolerability. However, since treatment monitoring was heterogeneous and etiologies, particularly cerebral anoxia, were distributed unequally among the groups, great caution is required when interpreting these data. A retrospective analysis taking into account possible combinations of anesthetics did not show any notable difference in outcome among the agents used alone or in association.15 There is also considerable uncertainty regarding the optimal extent of EEG suppression 15,57 and the optimal length of treatment. It is advisable to continuously monitor patients with EEG during treatment. A target of burst-suppression having an interburst interval of about 10 seconds, maintained for 24–36 hours, followed by progressive tapering over 12–48 hours represents a practical option.
Beyond the Lines and Prognosis
Other anesthetics, such as ketamine, an NMDA antagonist,69 or isoflurane,70 an inhalation anesthetic, represent options for extremely refractory SE, which is classically encountered in young patients with documented or presumed encephalitis,13 but are not used routinely. In the category of rescue drugs, one should also not forget LEV,41 as well as topiramate,71 which exerts its action through AMPA-receptor inhibition as well as ionchannel modulation.59
Short-term mortality of SE has been between 7 and 39% in epidemiological studies,9–11,72,73 varying according to different underlying etiologies, ages, and, in some studies, ethnicities. A long-term (10-year) study described a cumulative mortality rate of 43%, excluding the subjects who died within 30 days. This represents a three-fold risk compared with the general population.74 Functional outcome has not been systematically assessed.
The most important outcome predictors found in multivariate analyses are etiology, older age, and extent of consciousness impairment before institution of treatment.72,75–77 Importantly, a symptomatic SE episode increases the risk of developing epilepsy by a factor of three compared with isolated symptomatic seizures.78 In general, specific lifethreatening etiologies also carry a higher mortality risk relative to acute symptomatic SE.76 Refractory SE also increases the likelihood of subsequent epilepsy79 as well as a worse functional outcome15 compared with an SE episode promptly controlled by treatment. The role of SE duration in determining short-term outcome is unclear. The survival advantage of those whose SE is controlled within an hour may reflect less severe etiologies, while beyond one-hour duration is not clearly associated with outcome.77
Conclusion
Although several pharmacological options are used in the treatment of SE, there is a substantial lack of comparative, high-level, evidencebased information. These concerns need to be investigated in welldesigned studies. Since independent SE outcome predictors (etiology, older age, extent of consciousness impairment) seem to determine prognosis more than its treatment, it appears that the best current strategy involves familiarity with and prompt activation of a logical protocol, always with attention to the words of Hippocrates and Galen: first do no harm.