Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune-mediated disorder of the peripheral nervous system, of unknown etiology.1 The condition usually presents as weakness in the arms and legs, balance/gait impairment, tingling, numbness, and loss of tendon reflexes.2–4 It can be relapsing or progressive and the available treatments have variable efficacy in different patients.5,6 The symptoms of CIDP cause considerable burden for patients and their families and can cause loss of function and diminished quality of life (QoL).1,5,7
CIDP occurs in individuals of all ages but is more prevalent in men and in people over 60 years of age.3,8 The condition has an estimated incidence of 1.6/100,000/year (1.9–7.7/100,000)9 and an estimated prevalence of up to 8.9/100,000 (depending on criteria used to define the disease).9 CIDP may be slightly more common in patients with diabetes, though this has not been confirmed in population based studies.9 The infrequency of the condition means that many physicians and even neurologists will rarely see a case and may have difficulty differentiating it from other neurological conditions. Correct diagnosis and appropriate treatment may consequently be delayed.10
There are multiple subtypes of CIDP and it may actually be a spectrum of conditions rather than a single disorder.11,12 The variable presentation and course of the disease can make it difficult to diagnose and causes difficulties in assessing clinical trial endpoints.5,13,14 In addition, there is currently a lack of any reliable biomarker for CIDP diagnosis and there is a substantial need for clearer diagnostic guidelines.6 Many different criteria for the diagnosis of CIDP have been published and contribute to the variable incidence and prevalence reported in different publications.5,6,13,14 Despite these challenges, CIDP is a treatable condition. Effective therapies are available and new ones are being developed. Prompt diagnosis and initiation of treatment early in the course of the neuropathy are critical to prevent irreversible disability.5
Current treatments for chronic inflammatory demyelinating polyneuropathy
Intravenous immunoglobulin
There are a number of therapeutic options for treating CIDP, each with advantages and disadvantages. Differing response to therapy, medical comorbidities, side effects, and logistical factors all contribute to treatment decisions for any individual patient.
One of the most commonly prescribed and effective treatments for CIDP is intravenous immunoglobulin (IVIg). This is a well-established treatment that is widely considered first-line therapy in CIDP in patients without contraindications.5,15–18
The mechanism by which IVIg works in CIDP is not fully understood but is thought to act through multiple mechanisms. It has effects on B cells and antibodies (e.g. anti-idiotype mechanisms against pathogenic antibodies), complement (inhibiting complement activation), macrophage activity (Fc receptor blockade of macrophage activation), cytokine secretion, and leukocyte migration.19 These actions diminish inflammation and demyelinating processes and consequently prevent disease progression.
A number of studies have demonstrated the efficacy of IVIg.15,16,18 The largest trial to assess the efficacy of IVIg in CIDP was the ICE (Intravenous Immune Globulin [10% caprylate-chromatography purified] for the Treatment of Chronic Inflammatory Demyelinating Polyradiculoneuropathy) study (ClinicalTrials.gov identifier: NTC00220740). This was a randomized, placebo-controlled, response-conditional crossover trial in which a population of 117 patients with CIDP received either IVIg (an initial IVIg-C loading dose of 2 g/Kg over 2–4 days, followed by 1 g/Kg over 1–2 days, every 3 weeks for up to 24 weeks) or placebo.20,21 During the first treatment period, 54% of patients treated with IVIg and 21% of placebo-treated patients showed an improvement in Inflammatory Neuropathy Cause and Treatment (INCAT) scores (p=0.0002). Patients whose increase in INCAT score was consistently ≥1 point greater than at baseline were eligible to enter the maintenance phase during which improvements in INCAT score were also seen with IVIg treatment. Decreased relapses occurred with IVIg during a 6-month follow-up extension phase. The most common adverse events with IVIg were headache, fever, chills, hypertension, rash, nausea, asthenia and administration site reactions.
There are limitations and side effects associated with use of IVIg in patients with CIDP. As IVIg levels fall after as little as 2 weeks, efficacy can wane.22 Some patients experience ‘wearing-off’ that can occur before their next scheduled IVIg treatment.22 Optimized or personalized IVIg regimens may be needed to reduce ‘wear-off’ and maintain efficacy.23,24 Other limitations of IVIg are related to logistical issues of intravenous administration. Dosing can take at least 2–4 hours to administer, must be dosed every 3–4 weeks, and requires the presence of a healthcare professional in the hospital, infusion center, or home.17 Common side effects associated with IVIg infusions include headache, fever and chills, mild nausea, backache, and less commonly, vasomotor and cardiovascular symptoms. Rash can occur and occasionally a more severe allergic reaction.25 More serious and life-threatening complications include venous and arterial thromboembolic events. Caution using IVIg, particularly with products with a sucrose stabilizer is recommended in patients with renal impairment. Because of the fluid volume, caution is also advised, in patients with congestive heart failure.26–29
Corticosteroids
Oral and intravenous corticosteroids, such as prednisone and intravenous methylprednisolone, have been widely used as first-line therapy in CIDP and were the cornerstone of treatment prior to the development of IVIg. Despite their widespread use in CIDP, there is very little high-quality evidence available supporting treatment with corticosteroids.18 Experience from non-double-blind studies and longstanding clinical practice suggests a beneficial effect of corticosteroids in CIDP. Most patients treated with corticosteroids for CIDP are maintained on a tapering dose of prednisone. Alternative regimens include weekly pulsed intravenous methylprednisolone. Long-term corticosteroid use in CIDP, as in other inflammatory diseases, is limited by serious side effects, such as hyperglycemia, hypertension, weight gain, gastrointestinal bleeding, increased infection risk, myocardial infarction, osteoporosis, sleep disturbance, and others.30–32 Given the risks of long-term corticosteroid use, most patients are maintained on steroid-sparing or the lowest-possible dose corticosteroid regimens.
Plasmapheresis
Plasmapheresis (also known as plasma exchange or PLEX) is another first-line treatment that has demonstrated benefits in CIDP in multiple placebo-controlled studies, although the effects can be short-lived.33–36 The mechanism of plasmapheresis efficacy in CIDP depends upon removal of pathogenic antibodies, making treatment at regular intervals necessary to maintain response. Patients typically require treatment frequency of once weekly to once monthly. Because plasmapheresis sometimes requires central vascular access, frequent treatment and specialized centers familiar with the procedure, it tends to be used where corticosteroids or IVIg are not sufficiently effective or cannot be tolerated.37,38 Some patients who respond initially to IVIg but later fail to respond adequately may improve on the combination of IVIg and plasmapheresis.39,40
Other treatments
A variety of other agents have been used as experimental treatments for CIDP, especially for refractory disease.41 Mycophenolate mofetil (Cellcept®, Genentech, South San Francisco, CA, US) has been assessed in several small non-randomized studies and is sometimes used as steroid-sparing therapy.37,42 Intravenous pulsed cyclophosphamide, has shown benefits in treating CIDP, including complete remission in 11/15 patients in one small study.43 High-dose cyclophosphamide (200 mg/kg), without stem cell rescue, also provided long term remissions and notable QoL improvements, in some patients refractory to standard treatment for CIDP.44,45 Cyclophosphamide has significant short- and long-term side effects, including infection risk, effects on fertility and long-term increased risk of malignancy, so it is typically reserved for only the most severe and refractory disease. There are no placebo-controlled studies, demonstrating benefit of rituximab, the anti-CD20 monoclonal in CIDP, but rituximab has been beneficial in patients with CIDP associated with hematologic disorders, and has been described to be effective in a number of case reports and series.46–49 Rituximab has also been reported to be effective in the recently described IgG4 paranodal antibody variants of CIDP, with neurofascin 155 and contactin 1 antibodies, which often respond poorly to IVIg.50,51 Randomized, double-blind, placebo-controlled studies of methotrexate, fingolimod and beta interferon did not show benefit beyond placebo in CIDP.52–54
Autologous hematopoietic stem cell transplantation is another experimental approach to treat CIDP41,55 and several case reports and case series have reported promising efficacy. In one study (n=6) long-term efficacy was sustained from 6–18 months after transplant.56 Another study (n=11), reported significant improvements in INCAT scores within 2–6 months after treatment that was sustained throughout follow-up with a manageable complication profile.57 Some patients despite initial improvement after autologous stem cell transplants, later relapse.58 There is significant morbidity and risk of death with hematopoietic stem cell transplantation, so this has been reserved as treatment for only the most refractory cases. There is currently an ongoing clinical trial to better assess efficacy of stem cell transplantation as a treatment in CIDP (ClinicalTrial.gov identifier: NCT00278629).
Subcutaneous immunoglobulin therapy
IVIg therapy has a number of limitations, particularly related to systemic side effects and problems with long-term intravenous access.25,59 In many patients, these factors diminish QoL and independence.59 Subcutaneous immunoglobulin (SCIg) is an alternative treatment option for patients with CIDP who have stabilized on IVIg, and has the potential to address some of these issues.59 SCIg is a home-based, self-administered route of Ig therapy. SCIg has been reported to have a favorable systemic side-effect profile and enables closer maintenance of stable serum IgG levels.
A series of small clinical studies and case reports have shown that patients with CIDP who have responded to IVIg can benefit from a switch to SCIg.60–64 In some series there is less ‘wearing-off’ and QoL measures were improved after switching from IVIg to SCIg. In addition, the adverse event profile of SCIg was generally more favorable than that of IVIg.61,62,65–67
The PATH trial
The PATH (phase 3 subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy) trial (ClinicalTrial.gov identifier: NCT01545076) has evaluated the use of a 20% SCIg formulation (IgPro20, Hizentra®, CSL Behring, Pennsylvania, United States) in CIDP.68 In this trial, 172 patients who were stabilized on IVIg and demonstrated to be IVIg dependent, were randomized to weekly high-dose SCIg (0.4 g/Kg equivalent to 2 mL/kg), low-dose SCIg (0.2 g/Kg equivalent to 1 mL/kg) or volume-matched placebo for 24 weeks. In the intention-to-treat analysis, the primary endpoint, proportions of patients who had no CIDP relapse or did not withdraw from the study, were 67% of the high-dose group, 61% of the low-dose group, and 37% of the placebo group (p=0.001 and p=0.007 for high- and low-dose versus placebo; Figure 1).68 Time to reach the primary endpoint was significantly longer for both high- and low-dose SCIg versus placebo (p=0.0005 and p=0.007, respectively; Figure 2) but the difference between doses was not significant (p=0.48).68 Absolute risk reductions for relapse/withdrawal were 30% for high-dose versus placebo (p=0.001), 25% for low-dose versus placebo (p=0.007) and 6% for high-dose versus low-dose (p=0.32).68
A sensitivity analysis of patients who relapsed only showed lower rates for both doses of SCIg-treated patients compared with placebo: 19% on 0.4 g/kg and 33% on 0.2 g/kg IgPro20 compared to 56% of patients on placebo. Both doses differed significantly from placebo, with no statistical difference between the 0.2 g/kg and 0.4 g/kg doses. For secondary outcomes, there were significant advantages for both high- and low-dose SCIg in terms of INCAT score (p<0.0001), Inflammatory Rasch-built overall disability scale (I-RODS) centile score (p=0.0002), grip strength in dominant and non-dominant hands (p=0.0223 and p=0.0026, respectively), and Medical Research Council (MRC) sum score (p=0.0026).68 The SCIg doses also showed improved QoL measures. Local reactions occurred at a higher frequency with SCIg doses compared with placebo (Table 1), were characterized primarily by erythema, and swelling, and tended to decrease over time. One patient experienced an acute allergic reaction. Based on the findings of the PATH study, SCIg was approved for maintenance treatment in CIDP after stabilization with IVIg in Europe and by the US Food and Drug Administration (FDA), in March 2018.69,70
Overall, the PATH trial demonstrated that SCIg is a reasonable treatment option for patients already stabilized on IVIg therapy and may be a good choice for patients with systemic side effects from IVIG, difficulty with venous access, and those who have logistical issues scheduling frequent intravenous infusions. Limitations of SCIg therapy relate to the risk of relapse with treatment switch, difficulty administering for patients with limited finger strength, and local injection site reactions.68,71,72
Comparative studies
Only a few studies have been performed comparing treatments for CIDP. Short term studies of 6 weeks, comparing IVIg and plasmapheresis and IVIg and prednisone have not shown significant differences, in efficacy or safety.73,74 CIDP though is a chronic disease and these short studies do not reflect the safety and efficacy over the course of the disease.
Two different corticosteroid regimens were compared in a cohort of patients with CIDP in the PREDICT (pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy) study (n=40).75 After 12 months, there was little difference in efficacy between the two treatments. At this point, a total of 16/24 (67%) patients receiving pulsed dexamethasone and 10/16 (63%) receiving prednisolone were in remission (odds ratio: 1.2, 95% confidence interval [CI] 0.3–4.4). It was concluded that pulsed high-dose dexamethasone could be considered as induction therapy. A follow-up of this study after 4.5 years found that cure or long-term remission was achieved in 10/39 (26%) of patients after either one or two courses of pulsed dexamethasone or daily prednisolone.76 Of the patients who were in remission after initial treatment, half had a relapse (median treatment-free interval 17.5 months versus 11 months, respectively). In 7/12 of patients (56%) who did not respond the diagnosis of CIDP was changed to another condition. This prompted the authors to suggest that in patients who fail to respond to these treatments, an alternative diagnosis should be considered.
Comparing intravenous immunoglobulin with intravenous corticosteroids
Few studies have directly compared IVIg with corticosteroids in CIDP treatment and the relative risks and benefits are consequently not well understood. In the IMC (Immunoglobulin Methylprednisolone for CIDP) study (n=45), during 6 months of treatment, more patients discontinued treatment whilst receiving intravenous methylprednisolone than IVIg (52% versus 13% relative risk: 0.54, 95% CI 0.34–0.87, p=0.0085).77 After adjustment for sex, age, disease duration, comorbidity, modified Rankin scale and Overall Neuropathy Limitations Scale (ONLS) scores at enrolment, and previous treatment with IVIg and steroids the difference was still significant. (odds ratio: 7.7, 95% CI 1.7–33.9, p=0.007). The frequency of adverse events was similar in each group (p=0.1606). During the 6-month treatment period, patients who failed to respond were given IVIg as secondary therapy.78
In the IMC study, after the treatment period ended and a median follow-up of 42 months, 28/32 (87.5%) patients treated with IVIg (either as primary or secondary therapy) had improved compared with 13/24 (54.2%) who received intravenous methylprednisolone (as primary or secondary therapy).77 This long-term follow-up also showed that a similar proportion of patients who were responsive to IVIg or intravenous methylprednisolone eventually relapsed after treatment discontinuation (24/28 [85.7%] for IVIg compared with 10/13 [76.9%] for intravenous methylprednisolone, p=0.659). However, the median time to relapse was significantly shorter with IVIg compared with intravenous methylprednisolone (median 4.5 months versus median 14 months [p=0.0126], Table 2). Survival curves of time to clinical deterioration in patients who responded to therapy are shown in Figure 3.
A post-hoc analysis of patient data from the IMC study found that a pure focal distribution of demyelination at baseline was associated with early deterioration following corticosteroid therapy.79 Among patients with early deterioration (n=7), 71% showed a pure focal distribution of demyelination compared with 29% with a non-focal distribution (n=26). In patients with non-early deterioration, however, 19% had pure focal and 81% had a non-focal distribution (Table 3). The analysis also showed that lesser sensory abnormalities may also be associated with a pure focal demyelination pattern. The results suggested that focal/non-focal demyelination patterns could be used predictively in guiding treatment but more investigation is needed.
Factors influencing the choice of therapy in CIDP
Short term studies have shown similar efficacy for initial therapy for CIDP, between IVIg, plasmapheresis, and high-dose corticosteroids. Factors to consider when choosing treatment for CIDP include symptom type and severity, disease progression, comorbid conditions, and life style issues. Patients who are refractory to one first-line therapy may respond to another first line treatment.37 It is the authors’ typical practice to start with IVIg as first-line treatment for CIDP, in the absence of significant contraindication to this therapy. Plasma exchange, corticosteroids, and other immune suppressing therapies are typically used in patients intolerant, or not adequately responding, to IVIg. SCIg is a reasonable option for patients who have stabilized with IVIg. Patients who may be appropriate candidates for SCIg include those with systemic side effects with IVIg, venous access problems, frequent wearing off with intravenous infusions, and logistical barriers to intravenous therapy.
Discussion
A number of effective therapies are available for the treatment of CIDP but still they have significant limitations and side effects. A total of 20% of patients remain refractory to standard therapy and require alternative approaches emphasizing the need for new treatment approaches.80 While IVIg is widely regarded as first line therapy for CIDP and is effective in many patients, it has limitations for maintenance therapy in some people. Plasmapheresis and corticosteroids offer reasonable alternative treatment options but have their own side effects and treatment limitations. Cyclophosphamide and autologous stem cell transplantation remain options for patients with severe and refractory disease.
Data from the PATH study support use of SCIg in patients who have been stabilized on IVIg, and offers a new treatment option for patients who have difficulty tolerating systemic side effects from IVIg or have issues with venous access or logistical access to intravenous infusions.
There remains a need for comparative studies of different agents for CIDP treatment, but few have been completed or are in progress. As new immunomodulating agents are developed for a range of autoimmune and inflammatory disease, we hope to see new therapeutic mechanisms in development. ⬛
Figure 1: Proportions (%) of patients reaching the primary outcome measure (no relapse or no withdrawal for other reason) in the PATH trial
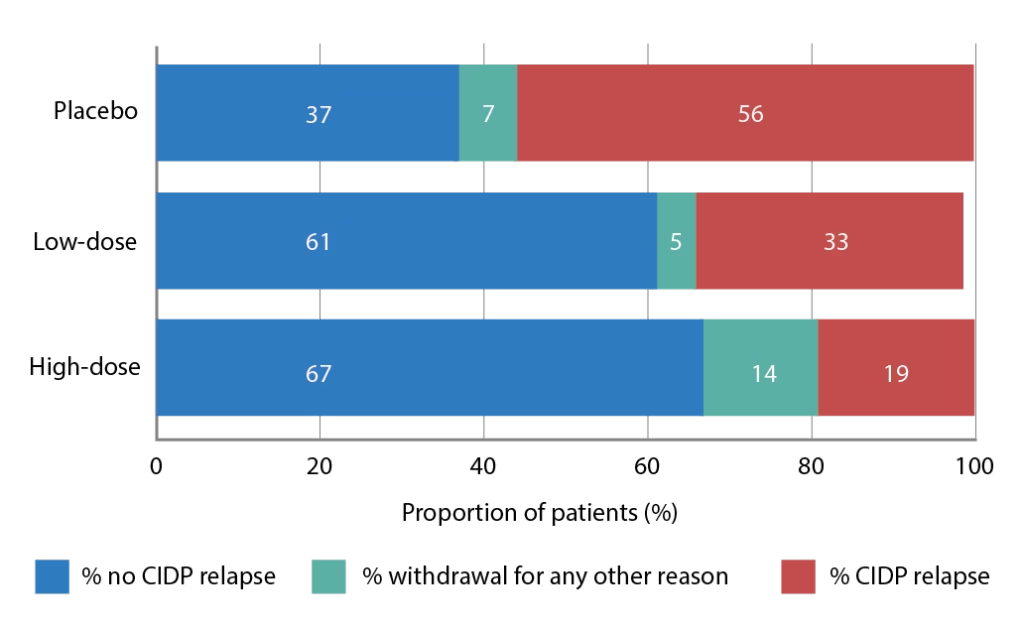
CIPD = chronic inflammatory demyelinating polyneuropathy; PATH = Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating
polyneuropathy trial.
Source: Replotted from supplementary data to van Schaik et al. 2018.68
Copyright permission from The Lancet Neurology.
Figure 2: Time to reach the primary endpoint during subcutaneous immunoglobulin or placebo treatment in the PATH trial
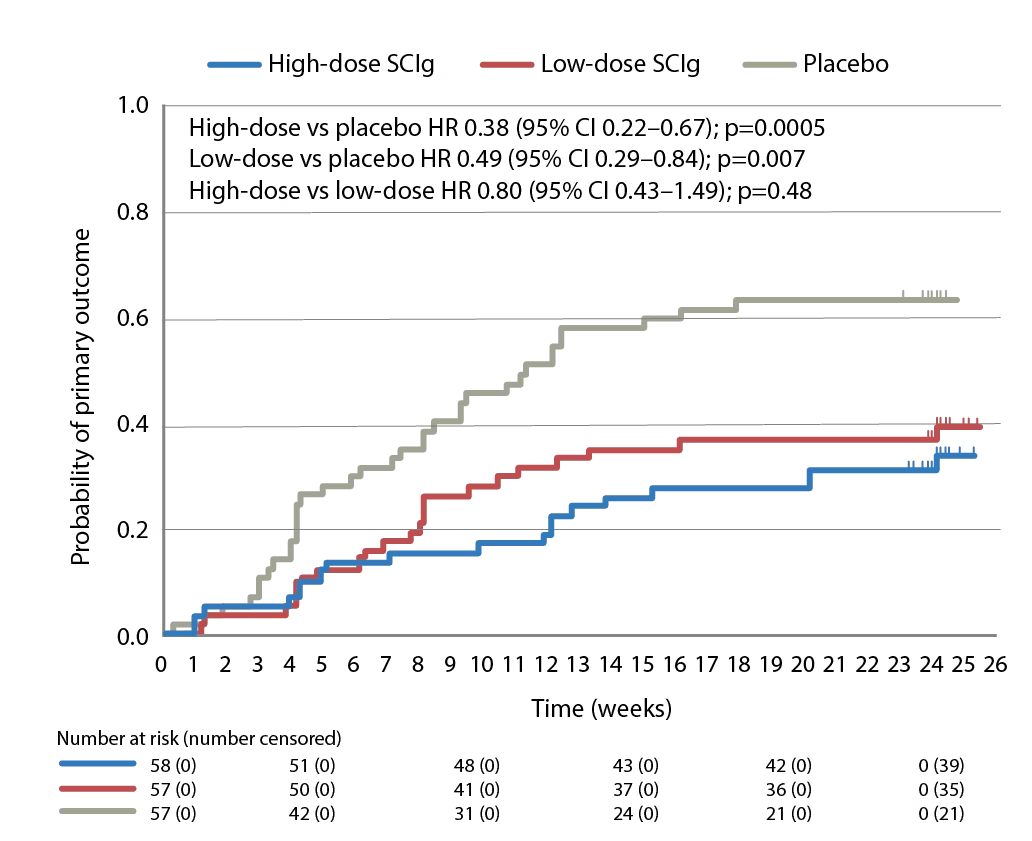
The primary endpoint in the PATH study was the proportion of patients who had no CIDP relapse or did withdraw from the study for any reason during the 24-week subcutaneous treatment period.
CI = confidence interval; CIDP = Chronic inflammatory demyelinating polyneuropathy; HR = hazard ratio; PATH = Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy trial. SCIg = subcutaneous immunoglobulin.
Source: van Schaik et al. 2018.68 Copyright permission from The Lancet Neurology.
Figure 3: Time to clinical deterioration after therapy discontinuation in patients with CIDP who responded to IVIg or IVMP treatment
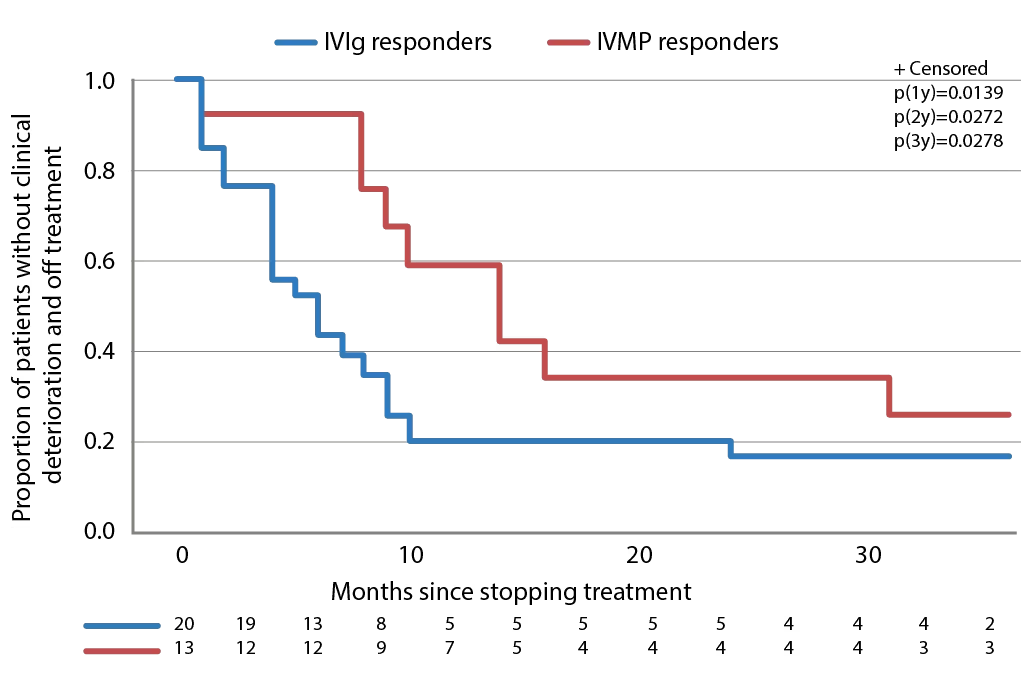
CIDP = Chronic inflammatory demyelinating polyneuropathy; IVIg = intravenous immunoglobulin; IVMP = intravenous methylprednisolone
Source: Used with permission from Nobile-Orazio et al. 2015.78
Copyright permission from BMJ.
Table 1: Adverse events in the PATH trial
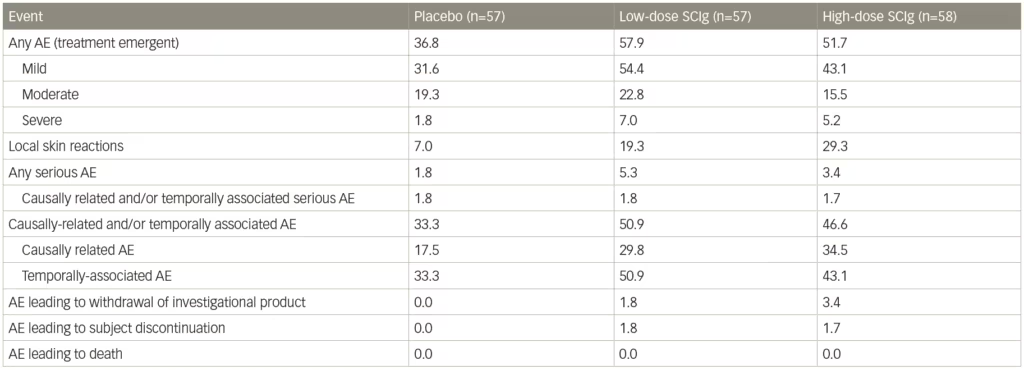
AE = adverse event; PATH = Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy trial; SCIg = subcutaneous immunoglobulin.
Source: supplementary data from van Schaik et al., 2018.68 Copyright permission from The Lancet Neurology.
Table 2: Deterioration and relapse after discontinuing a 6-month course of methylprednisolone treatment in a study of patients with chronic inflammatory demyelinating polyneuropathy
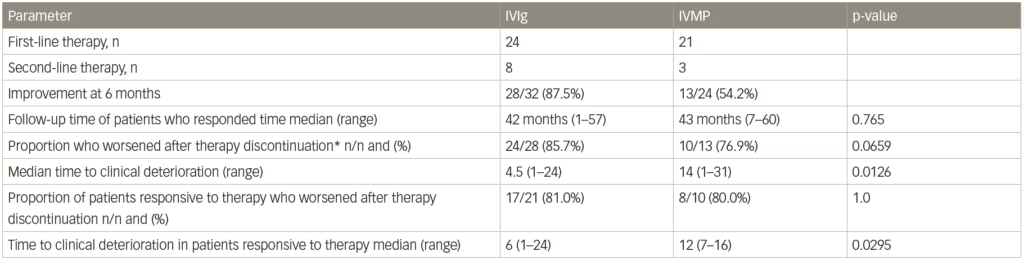
*Includes two patients who retired 1 and 7 months, and two who died 1 and 2 months after the last scheduled therapy (three after IVIg, one after IVMP).
IVIg = intravenous immunoglobulin; IVMP = intravenous methylprednisolone.
Source: Adapted from Nobile-Orazio et al. 2015.78
Table 3: Baseline electrophysiological and clinical parameters associated with early or non-early deterioration after corticosteroid treatment of chronic inflammatory demyelinating polyneuropathy
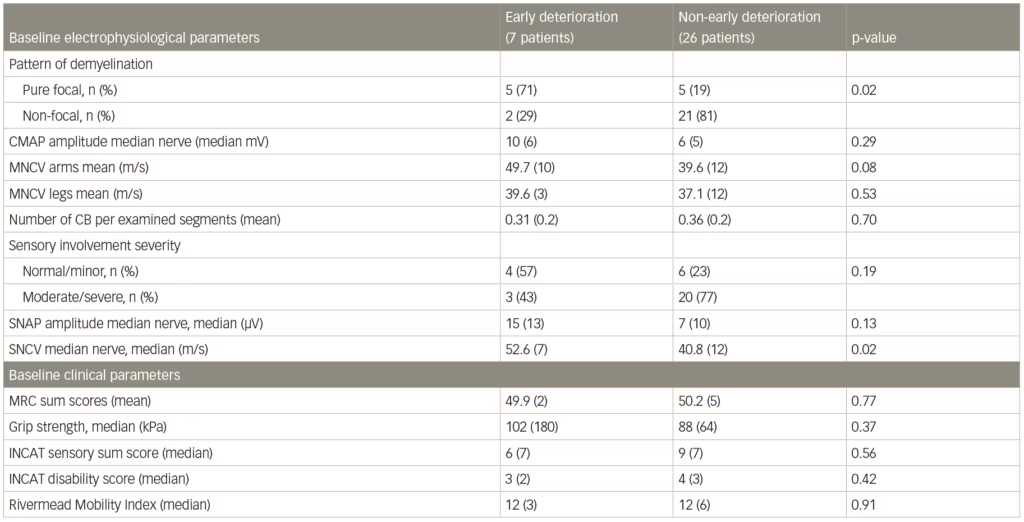
CB = conduction block (definite and probable summated); INCAT = inflammatory neuropathy cause and treatment; MNCV = motor nerve conduction velocity; MRC = Medical Research Council; SNAP = sensory nerve action potential; SNCV = sensory nerve conduction velocity.
Source: Eftimov et al. 2014.79