Myoclonus is defined as a sudden, brief, lightning-like muscle contraction.1 It was first described by Friedreich in 1881 when he detailed sharp jerks involving the bulk of a full muscle without marked limb or joint movement and called it paramyoclonus multiplex.2 Myoclonus may be described as either positive myoclonus (increase in contraction activity) or negative myoclonus (inhibition of contraction activity).1 This particular movement disorder can be quite difficult for the general practitioner, neurologist and movement disorder specialist to evaluate owing to its numerous aetiologies and a multitude of potential tests for proper diagnosis. The purpose of this article is to assist the clinician in understanding the aetiologies of myoclonus, as well as to serve as a guide in the diagnostic workup and treatment of myoclonus.
Classification of myoclonus
Myoclonus classification is based on multiple characteristics, most commonly including aetiology, anatomical distribution and physiology.3 It is important to consider all of these classification schemas when trying to decide diagnostic workup and treatment options. The article will briefly discuss physiological and body distribution classifications, as these tend to be most helpful in the diagnosis and treatment of myoclonus.
Body distribution classification
Myoclonus may be classified as focal, multifocal, segmental or generalized.4 Focal myoclonus is defined as the jerking of one body part. Multifocal myoclonus is seen in multiple noncontiguous body parts but is typically not synchronous.5 Segmental myoclonus is defined as rapid, jerk-like contractions in a group of muscles that are supplied by one or more continuous segments of the spinal cord or brainstem.6 Generalized myoclonus refers to jerk-like contractions of many body segments, usually in a synchronous fashion.5 Generalized myoclonus may be differentiated from multifocal myoclonus by a wider distribution of contractions in most or all body segments. Although body distribution is a helpful way to clinically classify myoclonus, body distribution alone is not enough to determine the underlying aetiology (and therefore treatment) of myoclonus. Thus, neurophysiological studies are helpful to further classify myoclonus.3
Neurophysiological classification
Neurophysiological testing for myoclonus helps to further localize the abnormal activation within the nervous system. Electroencephalogram (EEG) is an essential part of the workup for myoclonus, as myoclonus can be epileptic and most commonly arises from cortical or cortical-subcortical physiology.7 Along with EEG, electromyography (EMG) is another important tool to help determine the physiological classification of myoclonus.3 By using the aforementioned neurophysiological testing procedures, one can classify myoclonus as cortical, cortical-subcortical, subcortical-nonsegmental, segmental and peripheral.
Cortical myoclonus
Cortical myoclonus, arising from hyperexcitability of the sensorimotor cortex,8 is the most common type of myoclonus seen in the clinical setting.4 This hyperexcitability can be localized to one specific brain region, or it can spread along the motor cortex, which in turn leads to more widespread cortical activation and therefore myoclonus in more body regions. The impulses can even spread across the corpus callosum, leading to bilateral muscle activation. Thus, cortical myoclonus involves various body parts and can be classified as focal, segmental or generalized. On neurophysiological testing of cortical myoclonus, EEG-EMG back averaging will show time-locked cortical discharges just preceding short-duration burst potentials on EMG that last <50 milliseconds.9 The causes of cortical myoclonus are numerous, but some more common causes include chronic post-hypoxic myoclonus (Lance-Adams syndrome), progressive myoclonic epilepsy syndromes, and neurodegenerative conditions such as Alzheimer’s disease, Creutzfeldt-Jakob disease and dementia with Lewy bodies.
Cortical-subcortical myoclonus
Cortical-subcortical myoclonus arises from excessive abnormal oscillations between cortical and subcortical structures.4 This type of myoclonus is most commonly seen in the myoclonic seizures of juvenile myoclonic epilepsy. Cortical-subcortical myoclonus may also be present in absence epilepsy. On EEG, generalized spike and wave discharges should correlate with the myoclonic jerks to confirm cortical-subcortical myoclonus.10 Cortical-subcortical myoclonus may be focal, segmental or generalized.
Subcortical-nonsegmental myoclonus
Subcortical-nonsegmental myoclonus contains more variability in its neurophysiological characteristics than cortical or cortical-subcortical myoclonus, owing to the multiple locations from which activity may arise.11 Patients with subcortical-nonsegmental myoclonus do not have a correlation between EEG activity and any myoclonic activity seen on EMG. The myoclonus duration on EMG tends to be longer than in cortical or cortical-subcortical myoclonus, at up to 200 milliseconds.11 Some common causes of subcortical-nonsegmental myoclonus include propriospinal myoclonus, the myoclonus in myoclonus-dystonia syndrome and opsoclonus-myoclonus syndrome.
Segmental myoclonus
Segmental myoclonus is characterized by a generator, or area of abnormal excitability, in the brainstem or spinal cord that produces movements in the muscles related to a particular segment or a few contiguous segments.11 Similarly to subcortical myoclonus, segmental myoclonus does not have EEG activity that correlates with muscle activity. EMG activity is typically rhythmic and has a variable duration, between 50 and 500 milliseconds.9 Palatal myoclonus (palatal tremor) is the most prominent example of segmental myoclonus.
Peripheral myoclonus
This category of myoclonus arises from the peripheral nerve itself and has the widest variability in findings on EMG. The most common form of peripheral myoclonus is hemifacial spasm, in which the abnormal discharges arise from the facial nerve (cranial nerve VII).
Clinical approach
As myoclonus has such a wide array of localizations and aetiologies, a structured clinical approach is useful to ensure that all potential aetiologies are investigated for each patient. Similarly to many neurological disorders, history and physical examination both play an important role in determining what ancillary testing is needed for a thorough yet directed workup. It is important to have a detailed and systematic manner of approaching each aspect to ensure that the differential diagnosis contains all possible aetiologies for each patient.
History
As with most other disorders in neurology, the patient’s history serves as the foundational starting point for determining the aetiological differential diagnosis. Important historical clues include: the age of onset (childhood, early adulthood or late adulthood), the chronicity of the symptoms (acute, subacute or chronic), symptom progression (rapidly progressive, slowly progressive or static), coinciding factors (new medications or events that happen around the start of the symptom), family history and other associated symptoms, such as dementia, paraesthesia, weakness or loss of consciousness episodes.12 Age of onset can be especially useful in narrowing the differential diagnosis. Adult-onset patients are less likely to have genetic disorders, which often present with early onset of myoclonus. Similarly, family history can often serve as a clue for the diagnosis of genetic myoclonic syndromes. Similar observations can be made with the chronicity and progression of symptoms. Subacute myoclonus may be associated with metabolic abnormalities such as liver or renal failure, but also with paraneoplastic or autoimmune aetiologies. Other coinciding factors, especially new medications and any toxic or metabolic insults temporally linked to the onset of myoclonus, can be very helpful in determining the aetiology. Table 1 shows a list of common medication classes that may cause myoclonus.4 Finally, one should ask about associated symptoms such as dementia (and the chronicity thereof); Creutzfeldt-Jakob disease often exhibits startle-induced myoclonus, and Alzheimer’s disease and other neurodegenerative processes can have associated myoclonus in middle to later stages of the degenerative process. Each of these pieces of information will allow the examiner to begin formulating a differential around which to build the workup.

Examination
When examining a patient with myoclonus, it is important to determine the body distribution (focal, multifocal, segmental or generalized) as well as the constancy of the myoclonus (intermittent, continuous or rhythmic). Myoclonus may also be stimulus-induced, so it is important to assess for this during the examination as well. This may be done by simply touching the area of potential concern, or in other cases by auditory stimulus such as clapping hands or sudden closing of a door.12 Not only does the examiner need to attempt to classify the myoclonus, but a comprehensive neurological examination should also be completed to look for other symptoms that may aid in the diagnosis or help guide testing. For example, a patient who has myoclonus and parkinsonism (rest tremor, bradykinesia and rigidity) may have a corticobasal syndrome. Similarly, a patient with myoclonic jerks and dystonic posturing may have myoclonus-dystonia syndrome. A complete neurological examination will, along with a thorough and directed history, serve to guide the clinician in further diagnostic testing.
Ancillary testing
Following the history and physical examination, ancillary testing is the next step in identifying possible aetiologies in patients with myoclonus. Basic laboratory testing, such as serum electrolytes, glucose, thyroid studies and renal and liver function tests, should be obtained for nearly all patients.13 Drug and toxin screens can be considered as well, especially in the emergency department, inpatient setting or for the acute onset of myoclonus. If signs or symptoms of infection are present, then lumbar puncture should be performed, and cerebrospinal fluid can be evaluated for the presence of infection markers.13 In patients with subacute the onset of myoclonus and concern for immune-mediated processes, cerebrospinal fluid should be tested for specific antibodies (often available in laboratory panels).13 Some of the most common antibodies associated with myoclonus include LGI1 and CASPR2, both of which also present with encephalopathy.14 In nearly all cases of myoclonus (when available at the facility), EEG with EMG leads on muscles of interest should be done in concert. This allows neurophysiological diagnosis, which can have significant treatment implications. EEG in the absence of EMG can be useful to diagnose myoclonic epilepsies and coinciding seizures. Magnetic resonance imaging of the brain should also be considered to evaluate for structural causes, especially for segmental myoclonus. If there is a concern for segmental or peripheral myoclonus, then the spinal cord and/or peripheral nervous system should be imaged as well. If basic laboratory testing and imaging do not reveal a diagnosis, then specific genetic testing or whole-exome sequencing can be considered.15
Treatment
Treatment of myoclonus differs vastly depending on the aetiology. In cases that are secondary to medication/drug exposure or toxic/metabolic abnormalities, removal of the offending drug(s) and/or correction of the metabolic abnormalities is the first step in treatment. Myoclonus may also be part of epilepsy, such as in juvenile myoclonic epilepsy, in which antiepileptic medication is beneficial for myoclonus and other types of seizures. Once treatable conditions of myoclonus have been exhausted, then the clinician may consider symptomatic treatment, especially when there is not a treatable cause for the myoclonus and when the myoclonus is functionally disabling for the patient.11 Before selecting a pharmacological treatment for myoclonus it is important to consider the neurophysiological classification of the myoclonus, as different medications can treat different subtypes. Table 2 shows the common medications used to treat the different classifications of myoclonus.3
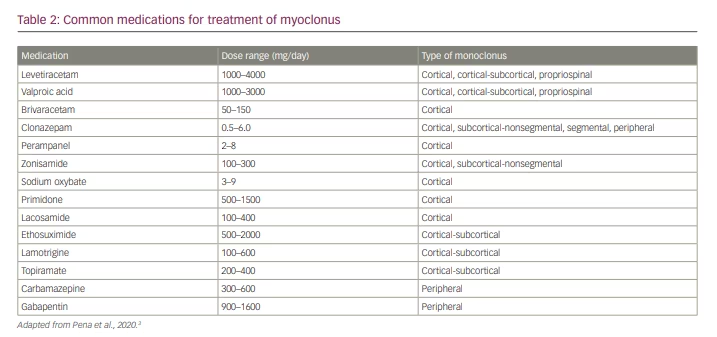
Cortical myoclonus may be effectively treated with medications that bind to synaptic vesicle protein 2a and are more commonly used to treat various types of epilepsy.16–18 Levetiracetam has evidence for the treatment of cortical myoclonus and is generally well tolerated with a low side effect profile.16 Piracetam has also shown efficacy in treating myoclonus in some studies but is only available in Europe.17 Some data from animal studies have shown that brivaracetam, the newest drug in the class, has symptomatic benefit in post-hypoxic myoclonus and some benefits in the myoclonic epilepsies, but further studies would be required before recommending brivaracetam for symptomatic treatment of general myoclonus.18
Valproic acid is another option for the treatment of cortical myoclonus and generally requires dosages ranging from 1200 to 2000 mg/day, but this medication typically works best in combination with other agents.19 Clonazepam may be used in cortical myoclonus, but, similarly to valproic acid, it shows the most benefit as adjunctive therapy with either levetiracetam or valproic acid.19 Other agents, such as zonisamide or primidone, are sometimes used to treat cortical myoclonus, but there is only limited evidence of benefit.20
For the treatment of cortical-subcortical myoclonus, especially that related to juvenile myoclonic epilepsy, valproic acid is considered to be the first-line therapy.21 Lamotrigine is another option, either as monotherapy or adjunctive therapy; however, it can worsen myoclonus in some patients, so close follow-up is warranted.21 Other adjunctive treatment options include clonazepam, ethosuximide and zonisamide.21 Most data regarding medications for the treatment of cortical-subcortical myoclonus come from studies of myoclonic epilepsies, which is the most frequent cause of cortical-subcortical myoclonus.
Subcortical-nonsegmental myoclonus is more challenging to treat owing to the large number of different underlying causative mechanisms. In this category overall, clonazepam seems to be the most effective treatment, especially in propriospinal myoclonus.22 Zonisamide may also be effective.22 If there is a clear structural lesion that is amenable to surgical removal, then removing the lesion could help to alleviate the symptoms.11 The opsoclonus-myoclonus syndrome, which is most often autoimmune or paraneoplastic in aetiology, also falls into this category. As with all autoimmune or paraneoplastic conditions, treatment of the underlying neoplasm or inflammatory condition will often improve the symptoms.11
Similarly to subcortical-nonsegmental myoclonus, segmental and peripheral myoclonus may have a broad differential, and focal lesions should be ruled out as part of treatment.11 For palatal myoclonus and hemifacial spasm, botulinum toxin injections can be beneficial.23,24 In some cases of hemifacial spasm, surgical decompression of cranial nerve VII may also be of benefit.25 In cases of spinal segmental myoclonus that are not due to a structural lesion or where the structural lesion cannot be safely removed, levetiracetam and clonazepam may be beneficial.26,27
Conclusion
Myoclonus is defined as a sudden, brief, lightning-like muscle contraction. Although myoclonus can be a normal physiological phenomenon, pathological myoclonus is a frequently encountered neurological symptom. The differential diagnosis for myoclonus is extremely broad, thus it can be difficult for a clinician to determine a targeted yet comprehensive diagnostic workup. A detailed neurological history and comprehensive physical examination can be combined with basic serum studies to exclude easily treatable conditions and to help guide further workup and ancillary testing. Classification by both body distribution and neurophysiology (using EEG and EMG) is vital to guide diagnostic testing and treatment. Treatment should be directed at any underlying cause of myoclonus where possible or feasible. When no other treatable cause is found, treatment for myoclonus should be targeted at specific neurophysiological classifications.