It has been nearly 160 years since Friedreich’s ataxia (FRDA) was clinically recognized and described1 and 25 years since the FXN gene was discovered.2 Despite this, there are still no approved therapies for FRDA.
FRDA is an autosomal, recessively inherited, neurodegenerative disease that typically presents in childhood and results in progressive gait and limb ataxia, with the extraneural features of hypertrophic cardiomyopathy, diabetes and scoliosis. At the cellular level, the genetic defect in FXN (first intronic GAA expansion on both alleles) results in deficiency of frataxin – a mitochondrial protein that plays a vital role in iron homeostasis, energy production via electron transport and response to oxidative stress – and affects tissues sensitive to mitochondrial dysfunction, including the brain and heart.3
An excellent review in 2020 by Zesiewicz and colleagues4 and an even more recent article by Pallardo et al.5 both discuss past and emerging therapies for FRDA, including antioxidants and mitochondrial-related agents, nuclear factor erythroid-derived 2-related factor 2 (NRF2) activators, deuterated polyunsaturated fatty acids, iron chelators, histone deacetylase inhibitors, transactivator of transcription-frataxin, interferon gamma (IFNγ), erythropoietin, resveratrol, gene therapy and antisense oligonucleotides.
The expert opinion of Zesiewicz et al. was that “While drug discovery has been challenging, new and exciting prospective treatments for FRDA are currently on the horizon, including pharmaceutical agents and gene therapy. Agents that enhance mitochondrial function, such as Nrf2 activators, deuterated polyunsaturated fatty acids, and catalytic antioxidants, as well as novel methods of frataxin augmentation and genetic modulation will hopefully provide treatment for this devastating disease.”4
However, a preclinically promising agent must navigate a number of potential stumbling blocks before it can progress to the finish line of US Food and Drug Administration (FDA) approval. Does it sufficiently distribute to the critical disease targets that modify the disease process? Is the clinical trial design adequate to demonstrate this? Do the benefits of treatment outweigh the risks?
To date, none of the preclinically promising agents investigated has yet overcome these challenges to unequivocally demonstrate clinically meaningful symptomatic or disease-modifying capability. Agents that are available outside of research protocols (vitamin E, coenzyme Q10, idebenone, deferiprone, riluzole, nicotinamide, IFNγ, erythropoietin, resveratrol, methylprednisolone, dimethyl fumarate, etravirine) continue to be used off-label by individual patients, despite a sometimes poor risk–benefit profile (e.g. for IFNγ6).
A detailed guide to the FRDA drug development pipeline can be found on the website of the Friedreich’s Ataxia Research Alliance (FARA) (www.curefa.org/pipeline), which is updated regularly. Drugs that have ‘fallen off’ the pipeline are summarized. Drugs in development that are in or nearing phase III completion are those that improve mitochondrial function and reduce oxidative stress (e.g. omaveloxolone7), closely followed by agents that modulate frataxin-controlled metabolic pathways (e.g. leriglitazone8). These are expected to show symptomatic benefit and possibly slow disease progression for a time.
The next wave of drug development will include agents that restore frataxin protein levels in the mitochondrion (e.g. transactivator of transcription-frataxin [TAT-frataxin]9), epigenetically allow DNA transcription to take place (e.g. Syn-TEF110) or replace or edit the mutated gene (e.g. AAV-based gene therapy11), bringing the field closer to a ‘cure’ – that is, stopping symptom progression or preventing onset altogether. However, reversing neural and extraneural damage that has already occurred – some during neural development12 – will remain a challenge for future research in cerebellar13–15 and spinal cord16 restoration.
In this review, we present an overview of the current therapies available and clinical evidence to support their use, and insights into developing treatments on the threshold of FDA approval. In addition, we discuss the symptoms of FRDA that can be managed, symptomatic medications that can be used off-label in FRDA and the progressive, late-stage complications of FRDA.
Diagnosis and the symptomatic management of patients with FRDA have been well defined in practice standards. Detailed clinical care guidelines are available on the FARA website (www.curefa.org/clinical-care-guidelines) and have recently been updated.
Diagnosis
FRDA is the most common cause of genetic ataxia in children aged 6–15 years, but it should be considered in all individuals with sporadic or recessive ataxia. It should also be considered in people with apparent dominant inheritance, due to the presence of undefined gait disorders in prior generations and the carrier frequency of 1 in 60–110 in populations of Western European descent (increasing to 1 in 11 in regions of Cyprus17) and the risk of pseudodominant presentation.18,19
The essential diagnostic criteria of Harding include onset before age 25 years, gait ataxia, limb ataxia, dysarthria, lower limb areflexia, extensor plantar responses and axonal sensory neuropathy.1 Other features occurring in more than 50% of most populations include decreased vibration, square wave jerks, dysphagia, weakness (foot dorsiflexion), amyotrophy (lower leg muscles), scoliosis, foot deformity, T-wave inversion on electrocardiography, hypertrophic cardiomyopathy on echocardiography and cervical cord atrophy on magnetic resonance imaging.
Regional genetic studies have expanded the phenotype associated with mutation of the FXN gene and the diagnostic confidence levels.20–23 Onset of symptoms after the age of 25 can be associated with retained reflexes and midline cerebellar atrophy, and such late-onset diseases can contribute to the diagnostic delay that can occur with an atypical presentation (e.g. onset with scoliosis or cardiac disease; phenotype of spastic ataxia or spastic paraparesis).24,25 Compound heterozygosity (one GAA expansion and one point mutation) can alter the severity and rate of progression of symptoms.26–28 A family with two point mutations was recently described, with a phenotype resembling Charcot–Marie–Tooth disease.29 A small number of patients who meet the criteria for FRDA have been found to have only one GAA expansion and no obvious point mutation, suggesting that promotor or other genomic changes might play a role.30 Finally, tissue mosaicism may explain some of the phenotypic variations seen with similar genetic testing results in blood.31,32
Genetic testing (www.ncbi.nlm.nih.gov/gtr) for the GAA expansion can be performed in 130 laboratories worldwide, with many of these laboratories also testing for point mutations.
Symptomatic management and follow-up
Large natural history studies in North America,33 Europe34 and Australia35 have provided key information on the types of symptoms and symptom progression seen in patients with FRDA, the most accurate ways to measure symptom severity and progression, and medications being used for symptom management. Practical guidelines for clinicians and patients should encourage:
- seeing a neurologist annually or sooner if unexpected symptoms/manifestations appear
- seeing a cardiologist annually or sooner if unexpected symptoms/manifestations appear
- seeing an orthopaedist annually to check the spine and feet or sooner if unexpected symptoms/manifestations appear
- signing up to the patient registry at www.curefa.org to receive notices about new research studies
- participating in the FRDA clinical outcome measures studies annually
- giving blood or other tissue samples for research
- annual blood testing to screen for diabetes (fasting blood sugar or haemoglobin A1c; urine sugar test)
- undergoing a baseline vision and hearing assessment
- being evaluated by a physical therapist and occupational therapist and following advice about home exercise36
- being evaluated by a speech therapist for speech and swallowing issues.37–39
Physical rehabilitation plays a key role in the management of patients with FRDA, at all ages and stages of disease.36 The main goals should be individualized and focus on stretching, core strengthening, balance and mobility (with or without assistive devices). Such an exercise programme can improve day-to-day independent functioning, reduce fatigue and pain, protect against muscle deconditioning, osteoporosis and joint contractures, and reduce the fall risk. Cardiac conditioning exercises need to be prescribed and monitored with respect to the patient’s underlying cardiac disease. Exercise programmes should not push patients to the point of pain or fatigue that lasts more than 1–2 hours after completion.
As there are no approved drug therapies for FRDA, many patients turn to vitamins, supplements and available agents in the pipeline (e.g. deferiprone, dimethyl fumarate, erythropoietin, etravirine, IFNγ1b, nicotinamide, resveratrol) to try to relieve or improve their symptoms. The proposed use of these unapproved agents must be guided by the attending neurologist. The most commonly used vitamins are the vitamin E family, coenzyme Q10 and idebenone. None of these has shown statistically significant benefits in clinical trials, but have been reported by some patients to improve fatigability and endurance.
The most recent Cochrane review40 noted that there is currently only low-quality evidence on antioxidants from two published small randomized controlled trials of idebenone41 or a combination of coenzyme Q10 and vitamin E,42 and that these neither supported nor refuted an effect on the neurological status of people with FRDA, as measured using a validated neurological rating scale. In addition, the review identified a large unpublished study of idebenone that reportedly failed to meet neurological endpoints and a trial of pioglitazone that has yet to be published, but is likely to influence quality assessments and conclusions on publication. The review noted that cardiac changes with these therapies were based on low- and very low-quality evidence of uncertain clinical significance. Low-quality evidence indicated that serious and non-serious adverse events were rare in both the antioxidant and placebo groups.
These findings suggest that relying on simple antioxidants alone will not be enough to alter the clinical course of FRDA, although they could help fatigability in some patients. Should a patient ask to try any of these unapproved agents, reasonable goals (e.g. how long to try, what benefits to look for) and safety monitoring should be discussed. Any symptomatic benefits (e.g. in fatigability, endurance) would be expected to be seen by 6 months.43
Common symptoms of FRDA (e.g. ataxia, upper motor neuron spasms, spasticity, contractures, weakness, fatigue, neuropathic or orthopaedic pain, restless legs, sleep disturbance, aids to mobility and fall prevention, dysarthria, dysphagia, vision loss, hearing loss, bladder or bowel dysfunction, sexual dysfunction, cardiac arrhythmias and heart failure, scoliosis and foot deformities, diabetes and mental health issues) are most effectively managed in a multidisciplinary setting, with rehabilitation medicine and with medications available for such symptoms in other, more common diseases.44
With better cardiac care,45–47 patients with FRDA are living longer and consequently experiencing significant disabilities related to chronic neurological decline. While the early stages of the disease primarily involve loss of ambulation48 and the need for accommodations at school or work,49 later stages bring an increasing need for assistance with activities of daily living, complications of being bedbound (e.g. infections, skin breakdown), nocturnal hypoxaemia, severe vision50 or hearing loss, and difficult-to-manage psychiatric issues (e.g. hallucinations, delusions).51 Palliative care, hospice care or long-term care placements (which can be difficult to find for relatively young people) may be sought by the patient’s family, requiring social work support.
It is important to listen to the patient and their family when developing a long-term treatment plan. Good resources for more information are FARA (www.curefa.org), the Friedreich’s Ataxia Parents’ Group listserv (contact Paul Konanz at pkonanz@comcast.net; email provided by permission) and the US National Ataxia Foundation (www.ataxia.org).
Access to clinical research
Collaborative translational and clinical research is necessary to assure the basic foundations for drug development in rare neurological disorders, and FRDA is fortunate to have many essential elements in place.
- The disease mechanisms are understood.52
- Candidate drugs have been developed.53
- A natural history baseline has been established.34,54
- Rating scales, quality-of-life measures and patient-reported outcome measures have been validated.49,55
- Biomarkers have been proposed.56
Research centres have been identified and patient registries opened (Collaborative Clinical Research Network in Friedreich’s Ataxia [CCRN in FA], www.curefa.org/network; and the Patient Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS), https://clinicaltrials.gov/ct2/show/NCT02069509).
The Friedreich’s ataxia clinical trial landscape
Since the discovery of the gene for FRDA, 55 registered interventional studies have been initiated and 39 completed.
None has shown statistically significant and clinically meaningful results. The question remains whether this is the result of the drug choice (e.g. poor penetrance, poor target engagement, poor biomarker response) or the study design (e.g. poorly powered, large placebo effect, too short, insensitive or not clinically meaningful outcome measures, study population too heterogeneous) – or any one of a number of other pitfalls in designing and conducting a clinical trial.
As FRDA is now considered a mixed degenerative and developmental disorder, some of its manifestations (related to abnormal development) are present at a very early age and possibly do not change much along the disease course. Recognizing which manifestations are developmental and which are degenerative is essential for properly choosing the outcome measures and biomarkers for a randomized clinical trial.57
EFACTS has addressed the issue of clinical trial design based on natural history data34 and the North American Collaborative Clinical Research Network will certainly add its insights to this. In a recent editorial in The Lancet Neurology on clinical trial design in FRDA, Savelieff and Feldman point out that: “Heterogeneity is a recurrent theme in FRDA; heterogeneity in clinical presentation, disease severity, tissue involvement, and FXN mutation render it essential to select the best and most robust outcome measures to track progression. This necessity is reinforced by the relative rarity of this disease and by its slowly progressive nature.”58
This reinforces the following issues.
- No single outcome measure can be ideal for a heterogeneous disease.
- Using patient subtypes to stratify a clinical trial to allow for heterogeneity will lower the number of participants and reduce the trial’s power.
- The correct balance between power and sample size can determine clinical trial success, as well as shortening the study duration and lowering its cost.
- Future trials in FRDA might benefit from matching the candidate drug to the subpopulation most likely to benefit. For instance, a trial testing a gene therapy might require a small cohort of young patients at an early stage of disease, while a trial assessing symptomatic treatment might require older, later-stage participants.
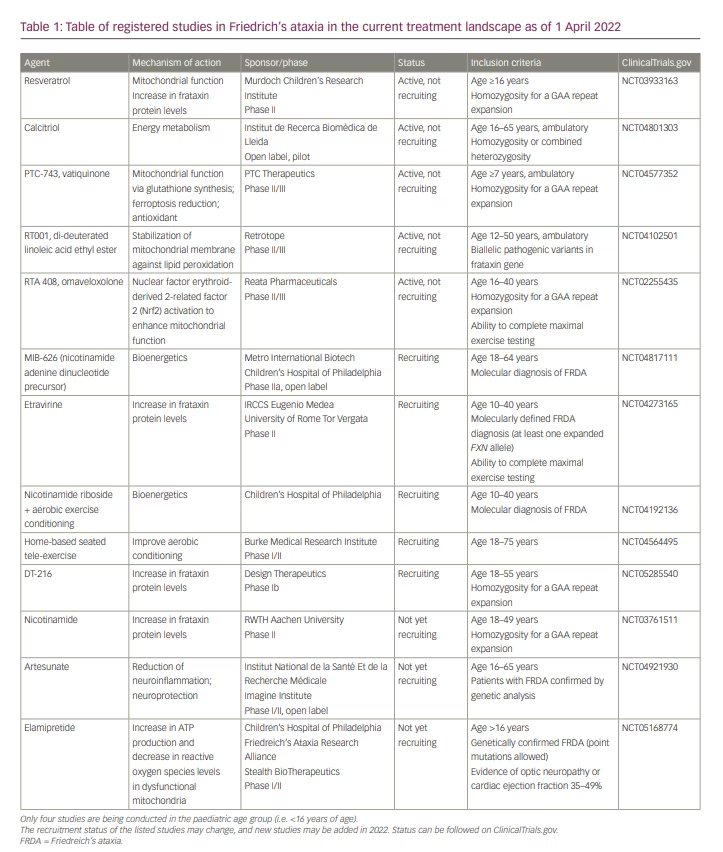
Table 1 shows registered studies in the current treatment landscape as of 1 April 2022.
Therapeutic programmes moving forward in 2022
Drugs that improve mitochondrial function
Reata Pharmaceuticals is preparing to submit a new drug application for omaveloxolone (an Nrf2 activator that improves mitochondrial function, restores the balance of reduction and oxidation, and reduces inflammation in models of FRDA) on the basis of data from its MOXIe Part 2 trial. This was an international, double-blind, randomized, placebo-controlled, parallel-group, registrational phase II trial conducted at 11 centres in the USA, Europe and Australia (ClinicalTrials.gov NCT02255435, EudraCT2015-002762-23).59
In the study, eligible patients, aged 16–40 years with genetically confirmed FRDA and a baseline modified FRDA Rating Scale (mFARS) score of 20–80, were randomized 1:1 to receive placebo or 150 mg/day omaveloxolone. The primary outcome was change from baseline in the mFARS score in the omaveloxolone versus the placebo group at
48 weeks. A total of 155 patients were screened, and 103 were randomly assigned to receive omaveloxolone (n=51) or placebo (n=52), with 40 patients in the omaveloxolone group and 42 in the placebo group analysed in the full analysis set.59
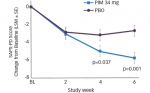
At week 48, patients in the omaveloxolone group had a mean decrease from baseline in mFARS score of –1.55 points compared with a mean increase of 0.85 points in the placebo group, giving a between-group difference of –2.40 points (p=0.014). Transient reversible increases in aminotransferase levels were seen with omaveloxolone, without increases in total bilirubin or other signs of liver injury. Headache, nausea and fatigue were also more common among patients receiving omaveloxolone.59
The difference between the treatment groups is the equivalent of turning the clock back 2 years in mFARS scores, although changes in secondary outcome measures (activities of daily living, frequency of falls, timed 25-foot walk, patient and clinician global impressions of change) that would add clinical meaningfulness did not reach statistical significance.
Data from a 1-year, open-label extension of the MOXIe study were previewed in a presentation to the International Parkinson and Movement Disorder Society in September 2021.60 The delayed-start results of patients transitioning from placebo to open-label drug supported the positive primary endpoint findings from the pivotal MOXIe Part 2 trial, with slowing of progression matching that seen in the original treatment cohort. Maintenance of the treatment effect in the original treatment group indicated a persistent effect of omaveloxolone on the disease course in FRDA. The safety profile in the MOXIe Extension was similar to that of Part 2, and no new safety signals have been identified to date.
Future trials with omaveloxolone may look at dosing, efficacy and safety in children younger than 16 years. In addition, trials of other agents will need to decide whether patients receiving omaveloxolone will be eligible for recruitment.
Two other drugs that aim to improve mitochondrial function and clinical outcomes in FRDA are being investigated in phase III trials and one in a phase II trial.
Retrotope is investigating the efficacy, long-term safety and tolerability of the deuterated polyunsaturated fatty acid RT001 in patients with FRDA (ClinicalTrials.gov NCT04102501). A total of 65 ambulatory participants, ages 12–50 years, were randomized to receive RT001 or placebo for 1 year, with a primary outcome measure of peak workload change from baseline to 11 months using cardiopulmonary exercise testing. There is no open-label extension. The study was due to be completed by December 2021. Top-line data analysis did not show a statistically significant benefit in the primary outcome measure for patients treated with RT001 versus placebo (manuscript in preparation). Further data analysis is ongoing.
PTC Therapeutics is conducting the MOVE-FA study, which is looking at the efficacy and safety of vatiquinone (ClinicalTrials.gov NCT04577352). The study sponsor anticipates recruiting 126 ambulatory participants aged 7 years and older, who will be given vatiquinone or placebo for 72 weeks during the placebo-controlled phase and for 24 weeks during an open-label extension, with a primary outcome measure of change from baseline in the mFARS score at Week 72. The study is anticipated to be complete by July 2023.
Minoryx Therapeutics is investigating the use of leriglitazone (also known as MIN-102), a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, on FRDA progression in the FRAMES study (ClinicalTrials.gov NCT03917225). This phase II proof-of-concept study has randomized 39 ambulatory participants aged 12–60 years to receive leriglitazone or placebo for 48 weeks, with a primary outcome measure of change from baseline in spinal cord area cervical segment C2–C3. Top-line data were released in December 2020, and showed improvement of relevant disease biomarkers in the brain and spinal cord and PPARγ engagement within the target range in all patients, as assessed by the relevant biomarker (adiponectin). Based on these results, Minoryx has indicated that it is advancing the programme, and will be meeting with regulatory agencies (the FDA and the European Medicines Agency) and planning a confirmatory study in FRDA.
Drugs that increase frataxin levels
It is anticipated that three new initiatives aimed not at mitochondrial function, but at increasing frataxin protein levels, will open enrolment for clinical trials in 2022.
Larimar Therapeutics has completed phase I studies of CTI-1601, a recombinant fusion protein intended to deliver human frataxin to the central nervous system, and is planning a phase II study.
Design Therapeutics is completing investigational new drug-enabling studies for its synthetic transcription elongation factor (Syn-TEF), with plans for a phase Ib study. Syn-TEF incorporates two distinct chemical moieties: programmable DNA binders that target desired genomic loci and ligands that engage the transcription elongation machinery. Polyamides are used as the DNA binders – in this case, selected for specificity for the GAA repeat – and are tethered to JQ1, which regulates RNA polymerase II pausing to promote transcription elongation. Initial studies have demonstrated that SynTEF1 can restore FXN expression in cell lines derived from patients with FRDA to the level observed in healthy cells.10
Recruitment is beginning for a phase Ia study evaluating DT-216 in adults with FRDA (ClinicalTrials.gov NCT05285540). This is a randomized, double-blind, placebo-controlled, single ascending dose study to evaluate the safety, tolerability, and pharmacokinetics and pharmacodynamics of intravenous DT-216. It will enrol 25 patients with FRDA aged 18–55 years with homozygous GAA repeat expansions.
Others
Companies with AAV gene therapy programmes (e.g. AavantiBio, Lacerta, Lexeo, Novartis, Pfizer, PTC, Takeda/StrideBio, Voyager/Neurocrine) targeting the central nervous system and/or heart are working to move preclinical programmes into first-in-human studies. AavantiBio’s product targets both the central nervous system and the heart, while Lexeo’s product targets just the heart.
Unfortunately, Exicure has terminated its FRDA programme due to issues with preclinical data. It had been completing investigational new drug-enabling studies, with plans for a phase Ib study, for its oligonucleotide-based spherical nucleic acid agent that focused on increasing frataxin gene expression and frataxin protein levels in FRDA.
Conclusion
This review is limited by the recently increased tempo of clinical and translational research in FRDA. In 2021 alone, more than 100 new publications about FRDA could be found on the PubMed database. More are likely to appear this year, and will hopefully answer some of the concerns addressed above.