Highlights
-
Limb–joint contractures may represent an important clinical clue of muscle dystrophies, as they limit the spectrum of the diagnosis assumptions.
-
Limb–girdle muscular dystrophies phenotype can rarely be a clinical presentation of retractile myopathies, except for non-specific Achilles tendon tightness.
-
Anti-PM/Scl antibody screening using line-blot assays and commercial antibody–panel testing is likely to detect false-positive antibodies.
-
Dysferlinopathies, calpainopathy and facioscapulohumeral muscular dystrophy are the conditions most frequently confused with inflammatory myopathies on muscle biopsy with prominent collections of inflammatory cells.
-
Before diagnosing inflammatory myopathies, muscle dystrophies with inflammatory characteristics on muscle biopsy must first be ruled out.
Muscle diseases with prominent limb–joint contractures (LJCs) are a subgroup of rare neuromuscular disorders with significant phenotype and genotype heterogeneity.1 LJCs and spine rigidity are of high diagnostic value and not exclusive to genetic myopathies. Ullrich congenital muscular dystrophy (MD), Bethlem myopathy and Emery–Dreifuss muscular dystrophies are the most common genetic causes of muscle diseases with LJCs. Inflammatory myopathies (IMs) can also cause contractures, typically following a painful inflammatory phase; they are generally not of genetic origin, although some people may have a genetic predisposition to developing these diseases. Prominent LJCs disproportionate to muscle weakness have occasionally been reported in the early stages of some limb–girdle muscular dystrophies (LGMDs), in particular LGMD type 1 (LGMDR1 or calpainopathy), dysferlinopathy and facioscapulohumeral muscular dystrophy. LGMDs are hereditary muscle diseases that are caused by genetic mutations and can manifest from childhood or adolescence, leading to progressive muscle weakness, particularly in the shoulders hip and leg muscles.1 Moreover, inflammatory infiltrate, which typically denotes IM, can be observed on muscle biopsy for LGMDs; as such, LGMDs may be confused with IMs.2 Consequently, definitive diagnosis is sometimes difficult to establish in patients with muscle involvement, especially when they are seen at a late stage, as this may signal both IM and MD.
Systemic sclerosis (SSc) is a rare condition that affects connective tissue and causes progressive fibrosis in various organs. Anti-PM/Scl antibodies are frequently observed in patients with SSc, with detection rates ranging from 3% to 11%.3 Furthermore, anti–PM/Scl antibodies have been shown to be associated with a high incidence of both myositis and LJCs in patients with SSc.4
For all of these reasons, we misdiagnosed a young girl with a 3-year history of painful muscular weakness with early and prominent LJCs and anti–PM/Scl antibodies as having juvenile scleromyositis, which is a subset of SSc in which patients with cutaneous features also present with myositis. In these patients, anti-PM/Scl antibodies are highly specific.
Persistently high creatine kinase (CK) levels after treatment, fatty replacement on muscle magnetic resonance imaging (MRI) and a dystrophic pattern on second muscle biopsy with absence of class 1 major histocompatibility complex (MHC) expression motivated us to reconsider a genetic disorder. Whole–exome sequencing revealed a novel mutation on the calpain 3 (CAPN3) gene, thus confirming the diagnosis of LGMDR1.
In this case study, we will discuss the difficulties in diagnosing genetic muscle disorders given the possibility of severe LJC in IM and of inflammatory infiltrate on muscle biopsy.
Case presentation
A 12-year-old girl from Tunisia, the first child of consanguineous parents with no family history of neuromuscular disorders or autoimmune diseases, was referred to our department for evaluation of gait disturbance. At the age of 9 years, she began to experience progressive weakness in her upper and lower proximal extremities, along with episodes of severe generalized muscle pain (measured at 80 mm on the visual analogue scale). Within the first year of the onset of her weakness, she began to walk on her toes. Upon neurological examination, muscle strength in her proximal upper extremities was 4/5 using the Medical Research Council scale, while muscle strength in her proximal lower extremities was 3/5. Mild weakness was also noted in her distal extremities. She had marked contractures in her elbows, long finger flexors and ankles (Figure 1) but no deformities in her feet or rigidity in her spine. There were no signs of scapular winging or calf hypertrophy, and skin examination revealed no rashes or papules and no visible evidence of Raynaud’s phenomenon. Her cardiovascular and respiratory examination findings were normal. CK levels ranged from 1,440 IU/L to 4,000 IU/L (7–20 times the normal level). Electromyography showed a myopathic pattern with spontaneous activity. Titres of anti-PM/Scl antibodies were positive. Serum studies for anti–nuclear antibodies, extractable nuclear antibodies and the anti–phospholipid antibodies were negative. A first biopsy of the deltoid muscle showed abnormal variation in fibre size with necrosis and cell intraparenchymal inflammatory infiltrate using haematoxylin and eosin stains. Chest computed tomography and capillaroscopy were within normal limits.
Initially, the patient was diagnosed with anti-PM/Scl antibody–associated myositis. Thus, she was started on daily 1.5 mg/kg oral prednisone tapered gradually and weekly 0.3 mg/kg oral methotrexate. At 12–month follow-up, there was no improvement in muscle weakness, and CK levels remained high (over 10 times the normal value). Skeletal muscle MRI in T1 and short inversion time inversion–recovery sequences showed no inflammatory signs and presence of fatty replacement involving quadriceps, adductors, biceps femoris, semitendinosus, semimembranosus and the gastrocnemius muscles, with a relative sparing of the gracilis and the anterolateral compartments of the legs. Consequently, a MD was highly suspected. A second quadriceps muscle biopsy revealed a dystrophic pattern with significantly increased connective–tissue volume, bimodal distribution of muscle fibres and persistent inflammatory infiltrate. Class 1 MHC was not expressed on muscle fibre in immunohistochemical stains (Figure 2). Whole-exome sequencing with xGen™ DNA Library Prep EZ kit (Integrated DNA Technologies Inc.; Coralville, IA, USA) confirmed the diagnosis of LGMDR1 (calpainopathy) based on a homozygous missense variant of the CAPN3 gene. Written informed consent for publication of clinical details and clinical images was obtained from the patient‘s parents.
Figure 1: Images of the patient showing contractures of finger flexors (A), ankles (B) and elbows (C)
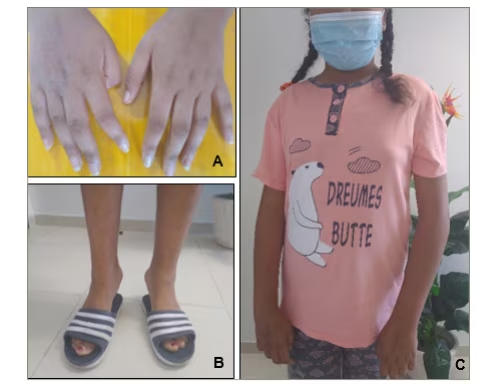
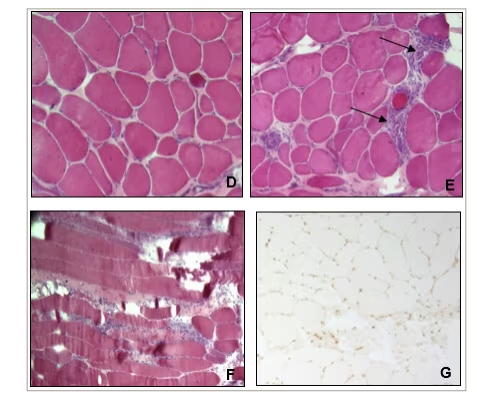
Figure 2: Muscle biopsy on haematoxylin and eosin stain
Muscle biopsy shows dystrophic changes with fibre necrosis, severe variability in fibre size and significant increase in connective tissue (D), associated with an inflammatory infiltrate (arrows) on transversal (E) and longitudinal sections (F). Immunohistochemical analysis are normal with absence of expression of class 1 major complex of histocompatibility on muscle fibres (G).
Discussion
LJCs are a very common feature in congenital and progressive myopathies, occurring either precociously or, by contrast, at late stages of the disease.1 When LJCs are the earliest and the most prominent clinical sign, they indicate retractile myopathies.1 Of retractile myopathies, the three most commonly diagnosed genetic diseases are: collagen VI-related myopathies (Ullrich congenital muscular distrophy and Bethlem myopathy), Emery–Dreifuss MDs (with mutations in the lamina A/C [LMNA], emerine and FHL1 genes) and selenoprotein 1 (SEPN1)–related myopathies.1,5 The LGMD phenotype is rarely a clinical presentation of retractile myopathies, except for cases of non–specific Achilles tendons tightness.1 Few cases of Becker MD,1 LGMD type 1F,6 facioscapulohumeral MD,7 BAG3–mutation-related myofibrillar myopathy8 and recessive mutations of the TTN gene9 have been reported.
In progressive MDs, LJCs usually appear at late stages of a disease and are associated with severe weakness and reduced mobility.4 This was the first factor that led us to error; our patient developed LJCs while she was still ambulant, when they usually occur when the disease is more advanced.
The second factor that misled us was the presence of anti-PM/Scl antibodies in our patient’s serum. These antibodies are detected in systemic autoimmune diseases such as polymyositis, dermatomyositis, SSc and overlap syndromes. According to one study, the specificity of anti-PM/Scl antibodies is reported to be 96.9% for SSc and 94.7% for CK elevation, and their presence is more common in SSc with myositis overlap syndromes.10,11
Both juvenile and adult cases of anti-PM/Scl antibody-associated myositis present with muscle weakness and myalgias, as well as with limited cutaneous SSc, Raynaud’s phenomenon, arthritis, mechanic’s hands and interstitdial lung disease.12,13 Juvenile cases are rare; about 45 cases have been reported in either a case report or in a larger series.14–16 In some cases, children with anti-PM/Scl antibodies may present with juvenile dermatomyositis years before developing SSc-like features.14 One study revealed that hand–joint contractures were significantly more common in patients with SSc who presented anti-PM/Scl antibody-associated SSc than in those who did not.17 Underlying causes for joint contractures in SSc include skin sclerosis, shortening of tendons due to peritendinous sclerosis, and joint deterioration.18 The course of paediatric–onset anti-PM/Scl antibody-associated myositis is chronic and displays a favourable outcome, with a good response to corticosteroids, and clinical remission. Flares of weakness or increasing CK levels after treatment are exceptional.16
Among the reasons that we ruled out a diagnosis of IM in our patient were its rarity, the fact that the most common myositis in children is dermatomyositis, and the fact that our patient lacked the characteristic skin signs.19 Additionally, the course of the patient’s condition was chronic, with severe pain, and all joints of the limbs were retracted, which is not typical in SSc. Furthermore, despite treatment, the patient showed no improvement and had persistently high CK levels, which did not indicate anti-PM/Scl antibody–associated myositis. A previous study evaluating the diagnostic yield of anti-PM/Scl antibodies using line-blot assays also showed inconsistent results, including false–positive cases,20 leading us to believe that our patient may have had a false–positive antibody detection.
The third factor that misled us was the presence of inflammatory infiltrate on muscle biopsy. Muscle inflammation is characteristic of IM but also occurs in MD; this represents a classic morphological pitfall. Dysferlinopathies, LGMDR1 and facioscapulohumeral MD are the conditions most frequently confused with IM in a muscle biopsy with prominent collections of inflammatory cells.2,21 In these cases, muscle imaging and muscle biopsy with immunohistochemical stains are of great interest. Class 1 MHC is not expressed on the sarcolemma of normal muscle fibres, and its presence is a marker of immune activation.22 Expression of class 1 MHC is a ubiquitous feature of IM and is used as a disease marker.23 Therefore, our data, including a positive result for anti-PM/Scl antibodies and presence of inflammatory infiltrates on a muscle biopsy, initially suggested a myositis diagnosis.
In accordance with evidence–based guidelines from the American Academy of Neurology,24 genetic causes should be suspected in our patient with an early onset age of muscular involvement. The pattern of muscle weakness suggested an LGMD, and consanguinity indicated an autosomal recessive mode of transmission. However, as it was a sporadic case, and consanguinity rates are high in Tunisia,25 it was also possible that it indicated autosomal dominant transmission. Furthermore, the dystrophic pattern observed, the lack of class 1 MHC expression, and the MRI results indicating selective involvement with fatty replacement and a lack of inflammation pointed towards a hereditary disorder. The presence of early and extensive muscle contractures led us to consider the diagnosis of retractile myopathy, with Emery–Dreifuss MD being the leading candidate. Therefore, further investigations were carried out to determine the underlying genetic cause of the patient’s condition. Finally, whole-exome sequencing indicated a mutation in the CAPN3 gene, confirming the diagnosis of LGMDR1.
Although rare in the Mediterranean area, and especially in Tunisia (0–24% of all recessive LGMDs),26,27 LGMDR1 (previously called LGMD2A) is a diagnosis that is compatible with our patient’s phenotype, histological pattern, imaging data and response to treatment. Typically, the disease occurs between the first or second decade of life, with progressive muscle weakness of the pelvic and scapular girdle muscles, and with severity varying from mild to severe.28 As mentioned above, early LJCs are rarely a clinical presentation of LGMDR1.1 Previous reports showed that patients with LGMDR1 may exhibit early contractures, especially in the ankles.29 It may also spread to affect the hips and the elbows.30 Muscle biopsy in individuals with LGMDR1 may reveal infiltrates consisting of eosinophils, macrophages and lymphocytes, that are often difficult to distinguish from those in IM.2,31,32 To our knowledge, the association of the early and prominent LJC phenotype and inflammatory pattern on muscle biopsy has never been reported with the CAPN3 gene.
The findings from our patient’s skeletal muscle MRI of fatty replacement involving various muscles in the legs, including the quadriceps, adductors, biceps femoris, semitendinosus and gastrocnemius, with relative sparing of the gracilis and anterolateral compartments, are in line with the pattern described by Barp et al. of fatty substitution predominantly involving the hip adductors and hamstrings in calpainopathies.33 On the other hand, the diffuse involvement of posterior and medial muscles reported by Caetano et al. was not seen in our patient.34 These findings suggested that our patient might have LGMDR1 but with a slightly different pattern of muscle involvement compared with previous reports.
LGMDR1 is due to mutations in the CAPN3 gene, which encodes calpain-3, a calcium-dependent protease that plays a role in cytoskeletal remodelling and membrane repair.35 Exons 1, 10, 5, 22 and 23 harbour most of the CAPN3 variants. Hotspot mutations for CAPN3 were reported, including c.550del for the European population and c.2120A>G for the Chinese population.36 Regarding domains of the calpain–3 protein, no additional risk was found in relation to mutations occurring in a particular region.36
Del1373C and del983A mutations in exons 11 and 17, respectively, were reported to be associated with LGMDR1 harbouring an early extensive LJC phenotype.30 Similarly, Landires et al. described a novel deletion in exon 3 of the CAPN3 gene (g.409_412del) with early severe contraction.37
A homozygous missense variant in exon 3 of CAPN3 c.386G>T; p.(Arg129Ile) linked to LGMDR1 was identified in our patient. This variant is not present in the Genome Aggregation Database38 and was predicted to be deleterious by different predictions tools: the Polymorphism Phenotyping v2 software found that it was probably damaging (0.978); the Sorting Intolerant From Tolerant algorithm classified it as deleterious (0); and the Combined Annotation-Dependent Depletion had a score of 35. The variant is heterozygous in the mother and father of the index patient. This mutation site involves the active catalytic site of the calpain cysteine protease (peptidase family C2) region, which may lead to the loss of the proteolytic activity resulting in a sarcopenic phenotype seen in LGMDR1.35
Our case study had some limitations. First, our initial approach of focusing on antibodies was erroneous, as our test results turned out to be false–positives. Second, we did not investigate the structural and functional consequences of the newly identified mutation. These limitations highlight the importance of careful and comprehensive diagnostic evaluation in the management of patients with suspected LGMDs, as well as the need for further research to fully understand the implications of novel genetic mutations in this disease.
In conclusion, this case report describes a novel mutation in the CAPN3 gene linked to early widespread LJCs and inflammatory infiltrate. Our findings emphasize the importance of considering calpainopathies in children presenting with myopathic involvement and early contractures. Additionally, our observations underline the significance of combining clinical findings, MRI and immunohistochemistry for diagnosing myopathies in low- and middle-income countries, where genetic testing may not be readily available. Finally, we suggest that, before making a diagnosis of IM, it is crucial to rule out MD with inflammatory characteristics on muscle biopsy, primarily by relying on clinical data. This approach is necessary to avoid further misdiagnoses and the inappropriate use of steroids and immunosuppressors in patients with genetic myopathies.