Myasthenia gravis (MG) is a relatively rare autoimmune disease, caused by an antibody-mediated blockade of neuromuscular transmission and resulting in skeletal muscle weakness. MG is characterised by fluctuating muscle weakness that worsens with activity and improves on resting. Over half of patients with MG initially present with ocular symptoms with or without generalised weakness.1–3 However, the disease progresses over weeks or months, with exacerbations and remissions.1 In the majority of patients, symptom onset to maximal weakness occurs within the first 2 years.3 While a number of immunosuppressive therapies are available, around 10% of patients with MG are termed refractory, experiencing frequent relapses upon lowering their immunotherapy or remaining clinically unstable on their current immunotherapeutic treatment regimen.4,5 This review article aims to discuss the current therapeutic options for MG and the potential of novel agents targeting the underlying pathogenesis of the disease.
Pathophysiology of myasthenia gravis
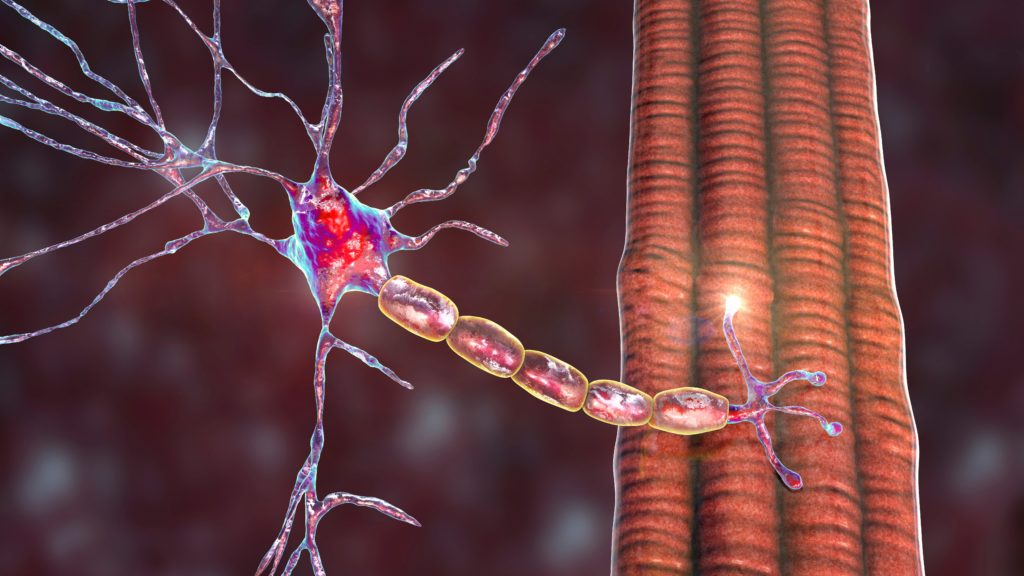
MG is the result of defective transmission between motor neurons and skeletal muscle. In around 80–90% of generalised patients, this is due to autoantibody formation against the acetylcholine receptor (AChR).6 The antibody interferes with neuromuscular transmission via blockade of receptor sites by steric hindrance, destruction of AChRs, and crosslinking of AChR, which causes increased turnover by endocytosis (from 5–6 days to 2.5 days), resulting in a loss of receptor density. This is followed by focal lysis of the post-junctional membrane by the terminal component of complement (Figure 1).6 Around 50–70% of generalised MG patients who do not produce antibodies against AChR are seropositive for antibodies against the muscle-specific receptor kinase (MuSK), which mediates the clustering of AChRs during synapse formation and is essential for the formation and maintenance of the neuromuscular junction (NMJ).7,8 In the remaining cases, termed seronegative, antibodies against AChRs and MuSK cannot be detected by available radioimmune assays.8 Clustered AChR-antibodies, which have demonstrated pathogenicity and have the ability to activate complement, have been detected in more than half of patients with previously seronegative generalised MG.9,10 Recently, immunoglobulin G (IgG)1 (and hence complement-fixing) antibodies against low-density lipoprotein receptor-related protein 4, which also is an essential component of the NMJ, have been found in a variable proportion of patients who were previously thought to be seronegative.11–13 Other autoantibodies to skeletal muscle proteins (titin, ryanodine receptor, myosin, actin, tropomyosin and troponin) have been found in the serum of MG patients, but their significance is not yet known.8,14

MG is considered a T-cell-dependent B-cell-mediated disease, in that CD4+ T-helper cells and T-regulatory cells facilitate the proliferation and differentiation of B-cells into AChR antibody-producing plasma cells, although they have additional different actions affecting the pathophysiology of MG.15 The thymus gland, the central organ in T-cell mediated immunity, is also an important factor in the pathogenesis of MG with AChR autoantibodies. It undergoes structural changes that make it like a tertiary lymphoid organ; however, its role has not been elucidated fully. It is the primary site of production of autoantibodies, and undergoes structural and functional changes in MG, and may be considered a tertiary lymphoid organ.16 Levels of thymic and peripheral blood CD4+CD25+ regulatory T-cells are reduced in patients with MG, and this is correlated with disease severity.17
The complement system and its role in myasthenia gravis
The complement system is a component of the innate immune defence against infection and is an important driver of inflammation in patients with MG who produce antibodies against AChR.18 It is strongly involved in the pathogenesis of MG.19 Excessive activation of complement can cause local and/or systemic inflammation, tissue damage and autoimmune disease. Immune complexes, indicative of a destructive autoimmune reaction involving the postsynaptic membrane in MG, were first detected in the 1970s.20 Complement deposits at the NMJ are a characteristic finding of MG, suggesting that AChR antibody induces muscle weakness by complement pathway activation, resulting in the formation of membrane attack complex (MAC), or terminal complement complex.19 The binding of complement factors to the AChR autoantibody results in the generation of a number of biologically active products, including anaphylactic peptides C3a and C5a, opsonic fragments C3b and C4b, and the MAC (which comprises C5b, C6, C7, C8 and C9). Any of these may contribute to the pathology of MG.18 The complement system is an attractive therapeutic target, as it is well characterised, has many natural inhibitors, and has a number of receptors that bind to activation fragments.21 However, treatment must impair complement deposition without detriment to the adaptive immune system.
Current therapeutic strategies in the management of myasthenia gravis
There are a number of current therapeutic strategies for MG, although no single regimen is appropriate for all patients, and treatment must be individualised.1 These include: acetylcholinesterase inhibitors, corticosteroids, other immunosuppressant drugs, thymectomy, and immunomodulatory therapies.
Acetylcholinesterase inhibitors
Acetylcholinesterase inhibitors have been used in MG for almost a century, and include pyridostigmine, neostigmine and ambenonium chloride.1 Pyridostigmine is recommended as part of the initial treatment in most patients with MG and is often used as maintenance therapy.22,23 They provide temporary (and often incomplete) relief of symptoms. Their efficacy is so clear that it would be ethically unjustifiable to perform a randomised controlled trial; although, their use does not affect disease progression from a pathological point of view.24 However, a proportion of patients (especially with purely ocular symptoms) would not require any other immunomodulatory treatment except the cholinesterase inhibitors.
Corticosteroids
Corticosteroids were the first immunosuppressant drugs to be used in MG and are recommended in patients whose symptoms persist despite treatment with pyridostigmine.22,23 The most commonly used corticosteroid in MG is prednisolone. However, corticosteroids are associated with serious side effects, especially with long-term usage.1,25 Nevertheless, as a result of their low cost and efficacy, corticosteroids remain the mainstay of treatment in MG around the world, which is supported by observational studies and expert opinion.26
Other immunosuppressant drugs
Azathioprine, which inhibits purine metabolism, as well as T- and B-cell production, is the steroid-sparing immunosuppressant drug that has been most used in MG.1 It improves weakness in most patients, but the benefit may not be apparent for 6–12 months.1 In a randomised controlled trial (n=34) of prednisolone plus azathioprine versus prednisolone plus placebo, there was no difference between the treatment groups until after 12 months.27 However, the same study showed that after 24–36 months, the relapse rate was lower with reduced steroid dosages, and there were longer remissions and fewer side effects in the azathioprine group. Its use is associated with hepatotoxicity and myelosuppression in around 15% and 9% of patients, respectively.28
Methotrexate is a commonly used alternative to azathioprine. There is some evidence for its effectiveness in MG, with one study showing that it is probably as effective as azathioprine as a steroid-sparing agent after 10 months.29 However, in a randomised controlled trial (n=50), methotrexate did not give any steroid-sparing benefit, and did not improve secondary measures of MG compared to placebo over 12 months.30
Cyclosporine is mainly used in patients in whom azathioprine is not tolerated and has been shown to improve muscle strength and reduce antibody levels, but it is also associated with side effects (35% discontinuation rate, of which 10% with renal toxicity), limiting its widespread use.31
Mycophenolate mofetil (MMF), which inhibits guanosine nucleotide synthesis and selectively inhibits activated T-cells, has a more favourable side effect profile than azathioprine.32 From 1998, when its use for MG was first reported, until 2008, MMF was considered an effective treatment option.33 A small study (n=5) found that MMF was effective and well tolerated as an adjunctive immunosuppressive therapy in patients with refractory MG.34 In 2008, two phase III studies did not demonstrate the superiority of MMF over placebo.35,36 However, in 2016, a retrospective cohort study found that discontinuation/marked reduction of MMF therapy may substantially increase the risk of MG exacerbation, supporting the commonly held view that MMF has a role to play in the maintenance of MG remission.37
Tacrolimus, which inhibits interleukin- (IL-)2, has shown some benefit in low doses and may be useful as a steroid-sparing agent, although evidence in support of its use is currently limited.38–40
It is clear from the above discussion that several immunosuppressive medications are moderately effective as steroid-sparing drugs in MG, but their onset of action may be delayed, the effect may not be sustained and very often their use is limited by severe side effects.
Thymectomy
Therapeutic ablation of the thymus gland (thymectomy) has been routinely performed in MG for more than 75 years. Thymectomy has been beneficial in numerous small case studies of carefully selected refractory MG patients.41–44 In addition, there are a number of clinically meaningful reports on large series of thymectomised patients but with widely varying rates of clinical improvement or remission.45–48 A systematic review found numerous methodologic flaws that prevented definite conclusions from being drawn regarding the benefits of thymectomy in patients with nonthymomatous MG.49 A randomised clinical trial (n=126) investigating the efficacy and safety of thymectomy was conducted in 2016.50 Patients were randomly assigned to extended transsternal thymectomy plus alternate-day prednisone or alternate-day prednisone alone. Patients who underwent thymectomy had a lower average Quantitative Myasthenia Gravis score over a 3-year period than those who received prednisone alone (6.15 versus 8.99, p<0.001). Patients in the thymectomy group also had a lower requirement for alternate-day prednisone. Fewer patients in the thymectomy group than in the prednisone-only group required immunosuppression with azathioprine (17% versus 48%, p<0.001) or were hospitalised for exacerbations (9% versus 37%, p<0.001). The incidence of treatment-associated complications did not differ significantly between the two groups, but patients in the thymectomy group had fewer treatment-associated symptoms related to immunosuppressive medications (p<0.001) and lower levels of distress related to symptoms (p=0.003). The investigators concluded that, over 3 years, thymectomy improved clinical outcomes and reduced the need for immunosuppressive therapy in patients with nonthymomatous MG.50
Further clinical trials are needed to ascertain the subset of patients who are most likely to benefit from the procedure. The effects of thymectomy are not immediate, and remissions may occur years later. It is also not known when it is the optimum time to perform thymectomy. However, thymectomy appears to be a reasonable therapeutic option in patients with generalised myasthenia and positive AChR antibodies and in any patient with a radiologically suspected thymoma.

Immunomodulatory therapies
Immunomodulatory therapy such as plasma exchange and intravenous immunoglobulin (IVIg) can be useful in acute MG exacerbations,51,52 and are widely used as the first-line treatments in myasthenic crisis. Both are useful techniques in the management of MG, but plasma exchange may need invasive central line insertions and can lead to potential complications. IVIg use can be limited due to supply issues, since this is a human-blood-derived product. These two therapies still remain the mainstay in patients with severe exacerbations causing bulbar muscle weakness, with or without ventilatory support.
In summary, there are several established and effective therapies for MG but most have not been tested using robust randomised controlled trials, or their efficacy has not been demonstrated in chronic or refractory MG. Attempts to perform clinical trials in MG have also been impaired by low patient recruitment or inconsistencies in clinical trial design, leading to the 2012 recommendations by the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America (MGFA) for future clinical trials. These included the use of a quantitative measures, such as the MG-Composite, that is weighted for clinical significance and incorporates patient-reported outcomes, consideration of alternative strategies for assessment of efficacy and safety, and development of predictive biomarkers.53
Practical management of myasthenia gravis
A proposed treatment algorithm is given in Figure 2. In patients with ocular or mild generalised myasthenia, the initial treatment is with cholinesterase inhibitors. If there is no significant response to cholinesterase inhibitors, most neurologists will commence treatment with steroids. The usual target dose of prednisolone is up to 0.5 mg/kg/day for pure ocular myasthenia and up to 1 mg/kg/day for generalised myasthenia, if using a daily regime. Some authors recommend alternate day dosing with a target of 0.75 mg/kg/alternate days for ocular and 1.5 mg/kg/alternate days for generalised patients.23 If symptoms are fully under control or the target dose is reached (whichever is earlier), the corticosteroids are usually continued for 2–3 months followed by gradual tapering to the smallest possible dose. The tapering regime may have to be tailored to the individual patient, but usually involves reducing by 10 mg/month until on 30 mg/day and thereafter by 5 mg/month until on 15 mg/day. Further reductions are usually in steps of 1–2 mg/month to the smallest possible maintenance dose, usually around 5–8 mg/day. If the maintenance dose of steroids is more than 7–10 mg of prednisolone/day or if the initial episode is very severe, most specialists commence steroid-sparing agents like azathioprine as mentioned earlier. Thymectomy should be considered as outlined above. There is increasing usage of early thymectomy by many MG experts, especially when they are AChR-antibody positive with generalised symptoms. Plasma exchange and IVIgs are usually reserved for patients who are at risk of, or in, myasthenic crisis.

Management of refractory myasthenia gravis
For patients in whom immunosuppressant therapy is not tolerated or not effective, there is no clear guidance on which therapy should be given (Table 1). Current MGFA guidelines recommend the use of azathioprine, cyclosporine, MMF, methotrexate, tacrolimus, chronic IVIg or plasma exchange, cyclophosphamide, or rituximab, but acknowledge that evidence for these therapies are lacking.22 Small studies of patients with refractory MG have demonstrated beneficial effects of high-dose cyclophosphamide, effectively ‘rebooting’ the immune system while leaving the haematopoietic precursors intact.54,55 However, these effects are often short-lived.54 Furthermore, cyclophosphamide is associated with a high risk of side effects. Isolated cases of autologous haematopoietic stem cell transplant, which resulted in long-term remission of refractory MG have also been reported.56,57
Rituximab is an IgG1 kappa monoclonal antibody that depletes B-cells by binding to their CD20 molecule and initiating complement-dependent cytolysis or antibody-dependent cell-mediated cytotoxicity.58 A number of small case studies/isolated case reports have suggested that rituximab is effective, well tolerated and produces durable responses in the management of refractory MG.4,59–66 In a recent retrospective case series (n=16), all patients achieved complete stable remission, pharmacologic remission or minimal manifestations of MG, and 44% of patients remained relapse-free with a mean follow-up of 47 months (range, 18–81) since the last rituximab treatment.66 Rituximab appears to be most beneficial in MuSK antibody-positive individuals.67 In a multicentre, blinded, review (n=199) 58% of anti-MuSK-positive patients with MG treated with rituximab reached the primary outcome of Myasthenia Gravis Status and Treatment Intensity level ≤2, compared with 16% of those not treated with rituximab This study provides Class IV evidence that for patients with anti-MuSK MG, rituximab increased the probability of a favourable outcome.68 While the evidence base for the use of rituximab continues to grow, and guidelines recommend that it may be considered in refractory disease, no consensus has yet been reached.22
Targeting the complement system
Targeting the complement system offers a useful therapeutic alterative for MG.6,18,69 Many complement inhibitors have been shown to reduce the incidence and severity of experimental autoimmune MG without causing substantial toxicity or alteration of immune function.69 The most successful strategy to date has been the use of an anti-C5 antibody.70,71 Blockade of the complement pathway at C5 halts the production of the pro-inflammatory and pro-thrombotic C5a and C5b molecules, which are important for inflammatory cell chemotaxis and activation of the MAC.72 Furthermore, this does not impair the immunoprotective and immunoregulatory functions of the proximal cascade.
The antibody-based C5 inhibitor eculizumab (Soliris®; Alexion Pharmaceuticals Inc., New Haven, CT, US) has been shown to be of potential use for MG treatment.73 In August 2017, the European Commission approved the extension of the current labelling indication for eculizumab to include the treatment of refractory generalised MG in adults who are anti-AChR antibody-positive.74 The US Food and Drug Administration (FDA) approval is for adults with generalised MG.75 Although the phase III Safety and Efficacy of Eculizumab in Refractory Generalized Myasthenia Gravis (REGAIN) study and its long-term open-label extension study (MG-302) did not achieve statistical significance in their primary endpoint of change from baseline in Myasthenia Gravis-Activities of Daily Living Profile (MG-ADL) total score, a secondary data analysis suggested beneficial effects of eculizumab in refractory MG patients in 18 of 22 pre-defined endpoints across four separate scales of disease severity.76
Complement inhibitors have the potential to cause opportunistic infections since they impair the function of the major host defence mechanism against invading pathogens.69 The labelling information for eculizumab includes the recommendation for meningococcal vaccination in patients with complement deficiencies.74 No meningococcal infections were reported in the REGAIN study.76 As mentioned above, the complement system is not significantly involved in the pathogenesis of MuSK-MG and hence the use of eculizumab is currently limited to AChR antibody-positive myasthenia patients.
Future therapeutic approaches
In addition to the limitations detailed previously, all current therapies for MG are non-specific and focus on the activity of T- and B-cells. However, novel immunotherapies are in clinical development, including a variety of T-cell directed monoclonal antibodies that block the intracellular cascade associated with T-cell activation, monoclonal antibodies directed against key B-cell molecules, and inhibitors of complement, cytokines and transmigration molecules. Early reports of the proteasome inhibitor bortezomib indicate promising efficacy in MG: it reduced anti-AChR antibody titers, inhibited damage to the postsynaptic muscle membrane, and resulted in clinical improvement.77 Belimumab, which binds to soluble B-cell activating factor and reduces B-cell activation and differentiation into antibody-producing plasma cells,78 has been evaluated in MG (NCT01480596).
Increasing evidence suggests that Th17 immune reactions play an important role in MG, and cytokines such as IL-17 and IL-6 may represent attractive therapeutic targets.79,80 Toclizumab, a humanised monoclonal antibody targeting the IL-6 receptor, has been found to be beneficial in cases of MG that did not respond to rituximab.81 Several human monoclonal antibodies against IL-17 are also in development, including brodalumab, ixekizumab and secukinumab, but these have not yet been tested in patients with MG.82
A potential adjunctive approach to MG is targeting muscle contractility. Tirasemtiv, a selective fast skeletal muscle troponin activator, binds to skeletal muscle troponin, thereby sensitising the muscle to calcium and ultimately improving muscle strength under submaximal stimulation. Results of a small (n=32), short-duration clinical trial suggest that tirasemtiv may improve muscle function in MG.83 Additional studies are needed to demonstrate efficacy in MG and determine optimal dosing. Short-term treatment with the β2 adrenergic agonist albuterol was shown to improve weakness in a mouse model of anti-MuSK MG.84 3,4-diaminopyridine, which enhances AChR release at the motor nerve terminal, has also shown promise in MuSK MG.85
Other potential approaches aim to target the anti-AChR autoimmune response and re-establish immune tolerance to the AChR. Possible approaches include administration of AChR or a portion of its sequence in a manner known to induce tolerance (e.g. oral or nasal); and disrupting the formation of the complex between major histocompatibility complex class II molecules, epitope peptide, T-cell receptor and CD4 molecule.6 However, these approaches have not progressed beyond the experimental stages.6
Summary and concluding remarks
The current standard of care in MG consists largely of generalised immunosuppression, which lacks specificity and selectivity. Currently approved therapies have a limited impact on refractory disease and around 10% of patients either fail to respond to treatment or suffer intolerable side effects. For such patients, there is a need for more aggressive treatment or treatment specifically directed to the underlying pathogenesis of the disease in order to prevent life-threatening crises, restore muscular strength and improve quality of life. Since there is a lack of clinical trial data, there is a need for registries to assess the effectiveness of the various therapeutic options. The MGFA has created a patient registry to promote research, treatment, advocacy and public awareness of MG (www.myasthenia.org).
The development of new, targeted therapies may help to improve quality of life in treatment-refractory patients. Disease heterogeneity in MG suggests that future therapeutic approaches should be tailored to MG subtype. Complement activation is an important contributor to the pathophysiology of MG and destruction of the NMJ; it is an attractive therapeutic target for the future. However, the utility of this approach will depend on the effect of complement inhibitors on the systemic immune system. Other novel therapeutic agents targeting the immunopathologic pathway underlying MG are also emerging. With the emergence of a number of potential therapies, there will be a need to develop biomarkers, which may help to effectively identify agents appropriate for later-phase testing.