Hereditary transthyretin amyloidosis (hATTR) is a progressive life-threatening disease that typically presents as progressive sensorimotor polyneuropathy, restrictive cardiomyopathy, or a combination of both.1 Patients with hATTR-associated polyneuropathy (hATTR-PN) typically require assistance walking after 5–6 years, and die within 7–10 years from the onset of neuropathy.2 However, clinical manifestations of amyloidosis can affect other organs as well, including kidneys, connective tissue, and eyes.3 Clinically, hATTR may be difficult to distinguish from the other major type of amyloidosis, light chain amyloidosis (AL), as prominent involvement of peripheral nerves and cardiomyopathy are a shared trait. Combined, hATTR and AL are almost 100 times more common than all other types of amyloidosis together.4 Rarely, these two types of amyloidosis may also present in the same patient.5
This review includes discussion of epidemiology, clinical features, diagnosis, and treatment of hATTR, with a focus on hATTR-PN.
Epidemiology
Worldwide prevalence of hATTR follows two patterns: a high density of the most common mutations in endemic areas, and sporadic and late onset in non-endemic areas. The highest prevalence of hATTR-PN is found in endemic areas in northern Portugal and Sweden, and specific regions of Japan, with most cases caused by the variant Val30Met.6 While initial reports of hATTR in Portuguese patients with Val30Met emphasized early onset of neuropathy, more recent worldwide studies show increased prevalence in the elderly, with more than 70% of patients diagnosed after the age of 60.4,7
In northern Portugal, the typical age of onset of hATTR is in the 30s, and by the age of 50, almost 80% of gene mutation carriers will have clinical manifestations of hATTR.8 In contrast, in Sweden, hATTR with the same transthyretin (TTR) mutation, Val30Met, manifests much later, and by the age of 60 only 22% of patients will become symptomatic.9 The onset of hATTR-PN may occur even after the age of 80.10 Most patients have heterozygous TTR mutations, while homozygous TTR mutations are associated with an earlier onset and more rapid progression.11,12 In addition to endemic areas, Val30Met can be seen in other ethnic groups and geographic regions worldwide, usually with a late onset after the age of 50.6 In the USA, the most common TTR mutation is Val122Ile, with an almost exclusively cardiac phenotype and disproportionately high prevalence among African–Americans.6 Rarely, hATTR with Val122Ile mutation may present with a dominant neuropathy phenotype.13 It has been estimated that this TTR mutation can be found in up to 3.4–3.9% of African–Americans, and there is often a late age of onset.14,15 Uncommonly, the Val122Ile variant is found in other ethnic groups as well.13,14 Other common TTR mutations in the USA include Thr60Ala with mixed phenotype and increased prevalence with Irish–Scottish ethnic background, and Val30Met with predominant neuropathic phenotype.6 The global prevalence of hATTR-PN has been estimated between 10,000 and 38,000.16
Wild type ATTR (wtATTR), previously also known as senile amyloidosis, is seen with accumulation of TTR amyloid deposits in the presence of normal TTR genotype, predominantly in the heart. WtATTR is much more common than hATTR, although its prevalence in the general population has not been established. It has been estimated that up to 13% of patients with heart failure with preserved ejection fraction may have wtATTR.17 It predominantly affects elderly men, with a median age at diagnosis of 78.6 years, with 18.5% of patients being women.18 While wtATTR is more common in the elderly, it has been known to affect patients as young as 47 years of age, and the median survival from diagnosis is only 3.6 years.19
Pathophysiology of nerve injury in hATTR
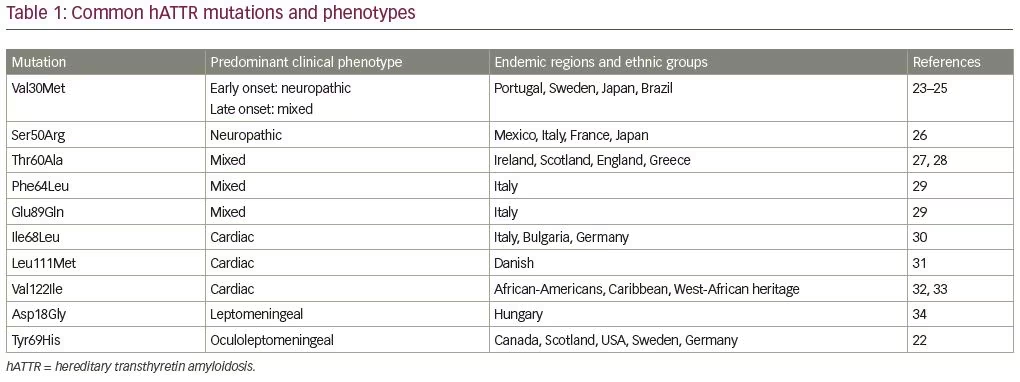
hATTR is an autosomal dominant hereditary disorder associated with more than 140 pathogenic TTR mutations leading to amyloid deposition, primarily affecting heart and peripheral nerves (Table 1).20–34 In its natural form, circulating TTR forms tetramers and serves as a shuttle protein for thyroxine and retinol-binding protein.35 Variant TTR from pathogenic mutations is more unstable leading to dissociation of TTR tetramers into monomers.35 Subsequently, amyloid deposits are formed from TTR monomers. Amyloid deposits often consist of both mutant and wild-type TTR, and it has been estimated that tissue deposits in most patients with hATTR may contain only 65–75% mutant TTR, while the rest is wild-type TTR (from the normal allele).20
Amyloid nerve injury is primarily caused by the accumulation of amyloid deposits in the endoneurium and nerve vasculature.20 Amyloid deposition starts around the endoneurial blood vessels with an early disruption of the blood–nerve barrier, which may facilitate buildup of amyloid in the endoneurial space.36 Patchy distribution of amyloid deposits in peripheral nerves may potentially explain some of false-negative nerve biopsies, as only a short nerve segment is examined. Teased-fiber studies demonstrate segmental demyelination in proximal nerve segments, while diffuse fiber loss with axonal degeneration is observed in distal nerve segments.37 Demyelination could be also partly attributed to injury of Schwann cells caused by amyloid deposits.36 Cumulative effects of multifocal amyloid lesions in proximal nerve segments might also contribute to the typical clinical picture of length-dependent neuropathy in hATTR.37
Clinical presentation and diagnosis of neuropathy associated with hATTR
Clinical presentation of hATTR-PN
hATTR-PN is a progressive disease with relentless worsening of symptoms and escalating disability.2 Clinical presentation and multisystemic manifestations of hATTR, to some degree, correlate with genotype; however, there may be a phenotype variability, even within the same family. While patients with hATTR typically present with a neuropathy-predominant or cardiac-predominant phenotype, most patients will have varying degrees of both nerve and cardiac amyloid deposition. Systemic deposition of amyloid results in a wide spectrum of symptoms reflecting multisystemic involvement, and amyloid neuropathy should be suspected in the presence of “red flag” symptoms, including bilateral carpal tunnel syndrome, restrictive cardiomyopathy, dysautonomia, and renal abnormalities.1,3
Late onset systemic amyloidosis can often be preceded by carpal tunnel syndrome for a decade or longer, due to amyloid deposits in tenosynovial tissue. In a study by Sperry et al., amyloid deposits were reported in tenosynovium of 10% of patients following carpal tunnel release, with a median age of 68 years, including seven patients with ATTR and two with AL (men >50 years, women >60 years).38
The most common phenotype of hATTR-PN is length-dependent symmetric painful axonal sensorimotor polyneuropathy with dysautonomia.39 Phenotype patterns of amyloid neuropathies and dysautonomia include generalized autonomic failure with painful large fiber polyneuropathy, generalized autonomic failure with polyneuropathy without pain, isolated generalized autonomic failure, polyneuropathy without generalized autonomic failure, and small fiber neuropathy with generalized autonomic failure.39 Clinical signs of dysautonomia may be absent at the onset of neuropathy, especially with late onset hATTR-PN after the age of 50 years.40 Electrodiagnostic testing in hATTR-PN typically shows axonal sensorimotor polyneuropathy with length-dependent involvement. Less common neuropathy phenotypes include demyelinating sensorimotor polyneuropathy (which may resemble chronic inflammatory demyelinating polyneuropathy), radiculopathy or radiculoplexopathy, motor neuropathy, and predominant upper extremity neuropathies (Table 2).22,41–47
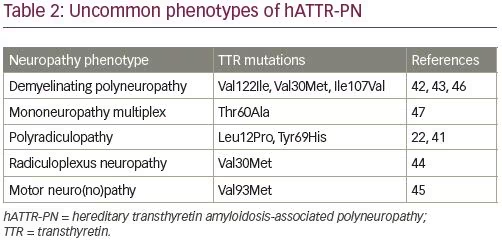
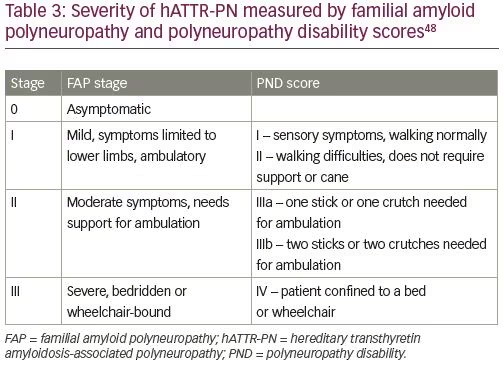
Clinical progression of hATTR-PN is usually described by staging based on Familial Amyloid Polyneuropathy (FAP) stage or Peripheral Neuropathy Disability (PND) score (Table 3).48 Dysautonomia is often among the most debilitating symptoms of hATTR, including orthostatic hypotension, syncope, and gastrointestinal dysmotility. In most patients with hATTR-PN orthostatic hypotension is primarily neurogenic, but the symptoms may be affected by the heart failure as well. Other manifestations of dysautonomia may include cardiac arrhythmias, neurogenic bladder, erectile dysfunction, and sweating abnormalities.49
Diagnosis of hATTR-PN
Diagnosis of hATTR is often delayed due to a wide spectrum of clinical presentations leading to unnecessary investigations, potentially harmful therapies, and delayed treatment.50 The presence of progressive neuropathy associated with multisystemic symptoms, including signs of cardiomyopathy, should raise a suspicion for possible hATTR-PN.1,3 While carpal tunnel syndrome often precedes the diagnosis of hATTR, its predictive diagnostic value is limited by its ubiquitous nature and high prevalence in the general population. In patients with known family history of hATTR, any new onset of peripheral neuropathy, dysautonomia, or cardiac symptoms should be considered as a possible manifestation of hATTR.1 Additionally, in patients without known family history, hATTR should be considered in the differential diagnosis of refractory progressive polyneuropathy, even in the absence of dysautonomia or cardiomyopathy. A wide spectrum of clinical presentations leads to a delay of diagnosis by an average of 4 years, often resulting in unnecessary testing and inappropriate treatment, with patients being evaluated by multiple physicians before diagnosis is made.50
The diagnosis of amyloidosis is established by the detection of amyloid deposits in tissue and accurate amyloid typing. Genetic testing is needed to confirm or exclude the diagnosis of hATTR (versus wtATTR). Easier access to genetic testing has greatly facilitated the diagnosis of hATTR, though we should not mistake non-amyloidogenic variants of TTR like Gly6Ser and Thr119Met, especially since Thr119 is associated with a lower risk of hATTR, even in the presence of pathogenic TTR alleles.21,51 An online database of TTR mutations is a helpful resource in the evaluation of patients with uncommon TTR variants (available at: http://amyloidosismutations.com/mut-attr.php).21
In patients with pathogenic TTR mutations it is preferable to confirm amyloid deposition in the tissue, which is a pathogenic event establishing that the patient has moved beyond the status of mutation carrier.1 The presence of amyloidosis in the tissue may be confirmed histopathologically, using biopsy or fat aspirate for example; or noninvasively, using bone scintigraphy or positron emission tomography (PET) scans.52,53 Bone scintigraphy tracers, technetium pyrophosphate (PYP), hydroxymethylene diphosphonate (HMDP), or diphosphono-1,2-propanodicarboxylic acid (DPD), allow an inexpensive, noninvasive method of demonstrating accumulation of TTR amyloid fibrils in cardiac tissue with high specificity and sensitivity;53,54 while PET scans are much more complex and expensive, and are typically not used in daily clinical practice. PET scans are also not specific for TTR amyloid deposits. Rarely, bone scintigraphy may be false-positive with deposition of non-TTR amyloid in the heart, primarily with AL.55 Additionally, paraproteinemia is not diagnostic of light chain amyloidosis, and monoclonal gammopathy of unknown significance is fairly common in ATTR.56 PET scans may allow non-invasive diagnosis of leptomeningeal amyloidosis using 11C-PiB or other amyloid-binding ligands.52 Cardiac magnetic resonance imaging may also demonstrate amyloid deposits, but cannot distinguish ATTR from AL.57
On histopathological specimens, the presence of amyloidosis is confirmed with various stains including Congo red, but such stains do not allow typing of the amyloid (ATTR versus AL). Technical inadequacy of biopsy sampling and processing, and patchy distribution of amyloidosis in biopsied tissue, may also limit the sensitivity of histopathologic examination. Immunotyping with anti-TTR or anti-light chain antibodies has limited sensitivity and specificity, and cannot distinguish hATTR from wtATTR.1 Most accurate amyloid typing on tissue specimens is obtained by mass spectrometry, which is the gold standard and is available in few centers.58,59 Nerve biopsy is now pursued less frequently, due to comorbidity associated with the procedure, easier access to genetic testing, possible false-negative biopsies, and the option to confirm amyloid deposition with lower morbidity procedures, including fat aspirates, gastrointestinal tract, or salivary gland biopsies.60,61 However, nerve biopsy may be very helpful in patients with progressive neuropathy when alternative causes of neuropathy are considered and may provide histopathological confirmation of amyloid deposition in the peripheral nerve. Amyloid deposition can be also demonstrated with skin punch biopsy.62
ATTR in other organs and tissues
Cardiac ATTR amyloidosis
Amyloid TTR cardiomyopathy (ATTR-CM) is the most common clinical manifestation of ATTR. Cardiac-predominant phenotype of hATTR is estimated to be at least four times as common as a neuropathy-predominant phenotype, although many patients have a mixed phenotype. Cardiac involvement is even more dominant with wtATTR, which almost exclusively involves the heart in most patients, with the exceptions of carpal tunnel syndrome, lumbar stenosis, and few patients with mild neuropathy.19,63 In patients with late onset hATTR-PN, cardiac amyloidosis may be present with limited clinical manifestations, at least in the early stages of the disease. The presence of ATTR-CM has a great impact on survival in ATTR and estimated median survival from diagnosis of ATTR-CM associated with Val122Ile mutation is 25.6 months, compared to 43 months with wtATTR.64
Amyloid deposition in the myocardium typically leads to restrictive cardiomyopathy with left ventricular hypertrophy, diastolic dysfunction, and heart failure with preserved ejection fraction.65 Diagnosis of ATTR-CM has been greatly facilitated by non-invasive scintigraphy demonstrating TTR amyloid deposition in the myocardium, and obviating the need for endomyocardial biopsies.54 In addition to increased thickness of the left ventricular wall, ATTR-CM is commonly associated with arrhythmias and cardiac conduction disturbances. Atrial fibrillation may affect almost two-thirds of patients with ATTR-CM, although it may be more common with wtATTR.18,19,65 A permanent pacemaker may be required in 25–36% of patients with hATTR and 43% with wtATTR.66,67 Patients with sustained ventricular tachycardia and high burden of non-sustained ventricular tachycardia may also benefit from implantation of a defibrillator.65
When compared with AL-CM, ATTR-CM is less commonly associated with kidney disease and patients are usually less symptomatic despite greater wall thickness and lower ejection fractions.65 Currently, tafamidis is the only medication approved in the USA for the treatment of ATTR-CM.68 Knockdown therapies are currently approved only for treatment of hATTR-PN, but the results of clinical studies suggest that inotersen and patisiran are also beneficial for cardiac manifestations of ATTR.69,70
ATTR in other organs and tissues
In hATTR, we can find amyloid deposition in many other organs and tissues, including the kidneys, eyes, meninges, and connective tissue. Most of the circulating TTR in the serum comes from the liver, but TTR is also produced in retinal cells and choroid plexus. Vitreous deposition of hATTR deposits may be the first clinical manifestation of hATTR. Ocular amyloid deposits were reported in up to 24% of examined patients with hATTR, more commonly in women (46% versus 15%).71 Clinical manifestations of ocular amyloidosis include vitreous amyloid, neurotrophic keratitis, glaucoma, and tortuous retinal vessels.71
Rarely, few TTR mutations may lead to leptomeningeal amyloid deposition, which can lead to seizures, cognitive decline, and brain hemorrhage.72 Other clinical manifestations of leptomeningeal amyloidosis may include ischemic stroke, hydrocephalus, encephalopathy, ataxia, spasticity, and radiculopathies. Leptomeningeal amyloid deposition in liver transplant recipients has uncertain clinical significance.52 Muscle amyloidosis has been reported in patients with hATTR and wtATTR-associated neuropathy and myopathy.73,74 Deposition of TTR amyloid fibrils in connective tissue, including muscle tendons, may precipitate carpal tunnel syndrome and biceps brachii ruptures.38,75 Currently available knockdown therapies for ATTR do not seem to affect mutant TTR production in the retina and choroid plexus and may not be effective for treatment of ocular and leptomeningeal ATTR manifestations.76
Other types of ATTR neuropathies
Wild type ATTR
While wtATTR primarily affects the heart, some patients may also develop peripheral neuropathy. The clinical course of neuropathy associated with wtATTR is typically indolent without significant dysautonomia.63,73,77,78 In addition to peripheral neuropathy, patients with wtATTR may also develop radiculopathies due to spinal stenosis associated with wtATTR.79
Acquired (post-domino liver transplantation) ATTR
Domino liver transplantation has been used in the treatment of hATTR, whereby the explanted liver from a liver-transplant recipient with hATTR is transplanted into a patient with liver failure, as such, the liver explant is otherwise functional, except for the production of mutant TTR. The rationale for domino transplantation was that patients with liver failure would have very short life expectancy without transplantation, and initial estimates suggested that only 0.4% recipients may develop amyloidosis from liver allograft with a delayed onset of acquired ATTR, many years after the transplantation.80 However, more recent studies showed that up to 48% of patients may eventually develop systemic TTR amyloid deposition, including at least 8% with an acquired ATTR-PN.81 Symptoms of acquired ATTR-PN start after a median of 7 years, although the onset has been reported as early as 2 months after transplantation.82 Rarely, acquired post-transplant ATTR-PN may have a fulminant course, as seen with rare Asn23Ser variant.83
Treatment of ATTR
If left untreated, hATTR-PN follows a progressive clinical course with a rapid functional decline and survival of 7–10 years.2 Until recently, the only available treatment options for hATTR included liver or liver–heart transplantation and off-label use of the TTR stabilizer, diflunisal, which both slow the progression.84,85 The TTR stabilizer, tafamidis, was approved in Europe, Japan, and Brazil in 2012 for the treatment of early and intermediate stages of hATTR-PN,86 and it was made available in the USA in 2019 only for treatment of ATTR-CM (both hereditary and wild type).68 Tafamidis is not approved for the treatment of ATTR-PN in USA at this time. Another TTR stabilizer, diflunisal, is a repurposed non-steroidal anti-inflammatory drug (NSAID) that has been available for treatment of arthritis in the USA since 1982. Recent studies showed promising results with blocking of TTR expression (knockdown), leading to US Food and Drug Administration (FDA) approval of the small interfering RNA (siRNA)-based patisiran, and the antisense oligonucleotide-based inotersen in 2018.87,88
The TTR stabilizers tafamidis and diflunisal reduce formation of amyloid fibrils by stabilizing the TTR molecule in its tetramer conformation, while patisiran and inotersen knock down the production of both mutant and wild-type TTR. Commonly used measures of efficacy of hATTR in clinical trials include various measures of quality of life, including the Norfolk Quality of Life–Diabetic Neuropathy (QOL-DN) and neuropathy severity scales, that are mostly based on the Neuropathy Impairment Score (NIS), including NIS-lower limbs (LL), NIS+7 and modified NIS+7.89
Knockdown of the TTR gene
Two knockdown treatments that were recently approved by the FDA, patisiran and inotersen, use sequence-driven cleavage to reduce levels of TTR mRNA in the liver and its protein product. These two medications, respectively, block the production of TTR by distinct mechanisms, with double-stranded siRNA and antisense oligonucleotides, but the main principle remains the cleavage of TTR mRNA through RNase H and RNA-induced silencing complex.90 Serum levels of TTR are reduced by 75–85% with knockdown therapies.
Treatment with inotersen and patisiran typically stabilizes symptoms, and some patients may even show improvement. Clinical studies have shown significant differences in measures of quality of life (Norfolk QOL-DN scale), neuropathy outcome scores (NIS +7/mNIS+7), and 10-minute walk tests in treated patients when compared with a placebo group.87,88 Furthermore, 36% and 50% of patients treated with inotersen, and 56% and 51% of patients treated with patisiran, had an improvement of neuropathy outcomes and quality of life at 15 and 18 months, respectively.87,88 Patisiran is given intravenously, whereas inotersen is given subcutaneously. Prior to patisiran infusions, patients need to be premedicated with corticosteroids, acetaminophen, and H1/H2 blockers.91 The use of inotersen is associated with the risk of potentially fatal thrombocytopenia and glomerulonephritis, and continuous monitoring is needed.88 Lowering of serum TTR level also reduces vitamin A level, so vitamin A supplementation is recommended with both TTR knockdown therapies. So far, there are no reports of major medical problems attributable to prolonged knockdown of TTR.
Another knockdown siRNA investigational treatment, revusiran, was abandoned after a study showed greater, predominantly cardiovascular, mortality in the treatment arm of a cardiomyopathy study (12.9% versus 3.0%), although subsequent analysis did not show any deleterious effects of revusiran on cardiac function.92 Non-ambulatory and terminal stage patients have been excluded from clinical studies and are unlikely to attain major benefits with such treatment, although their quality of life may improve. At this time, knockdown therapies are approved only for the treatment of hATTR-PN, and initial studies suggest benefits for hATTR-CM, as well. Studies investigating the use of knockdown therapies in wtATTR are in progress.
TTR stabilization
In its natural form, circulating TTR forms tetramers and amyloid deposits are formed from TTR monomers following destabilization and dissociation of TTR tetramers. Dissociation of TTR tetramers is the rate-limiting step of amyloidogenesis, and slowing of dissociation leads to reduction of clinical symptoms.93 Pathogenic TTR variants are associated with greater rates of TTR tetramer destabilization. Several compounds stabilizing TTR tetramers have been used in treatment of ATTR, including tafamidis, diflunisal, and most recently, experimental medication AG10.
Tafamidis has not been approved for the treatment of hATTR-PN in the USA after initial neuropathy studies, due to limited benefits, but it was approved for treatment of patients with early-stage hATTR-PN (stage I) in Europe, Japan, and Latin America (Table 4). A clinical trial enrolling patients with a Val30Met mutation showed that at 18 months, treatment with tafamidis was associated with a greater chance of slower progression (NIS-LL responders defined as having decline of <2 points) when compared with placebo (60% versus 38%), but the co-primary endpoints of the study were not met (NIS-LL and Norfolk Quality of Life [TQOL] scales).94 Long-term studies with tafamidis have shown slowing of progression in patients with Val30Met and other TTR mutations.95,96 One-third of the patients treated with tafamidis may still do well after 3 years of treatment.95 Early treatment often delays the onset of progression of neuropathy symptoms.97 Additionally, tafamidis also reduced the risk of mortality in patients with early and late onset ATTR-PN Val30Met compared with untreated patients.23 In patients with ATTR-CM (both hATTR and wtATTR), tafamidis was shown to reduce all-cause mortality by 13% and the risk of cardiovascular hospitalizations by 31%, with reduced rate of worsening on a 6-minute walk test (p<0.001).68 The dose of tafamidis in ATTR-CM trial was four times higher than previously used in treatment of ATTR-PN.68,94
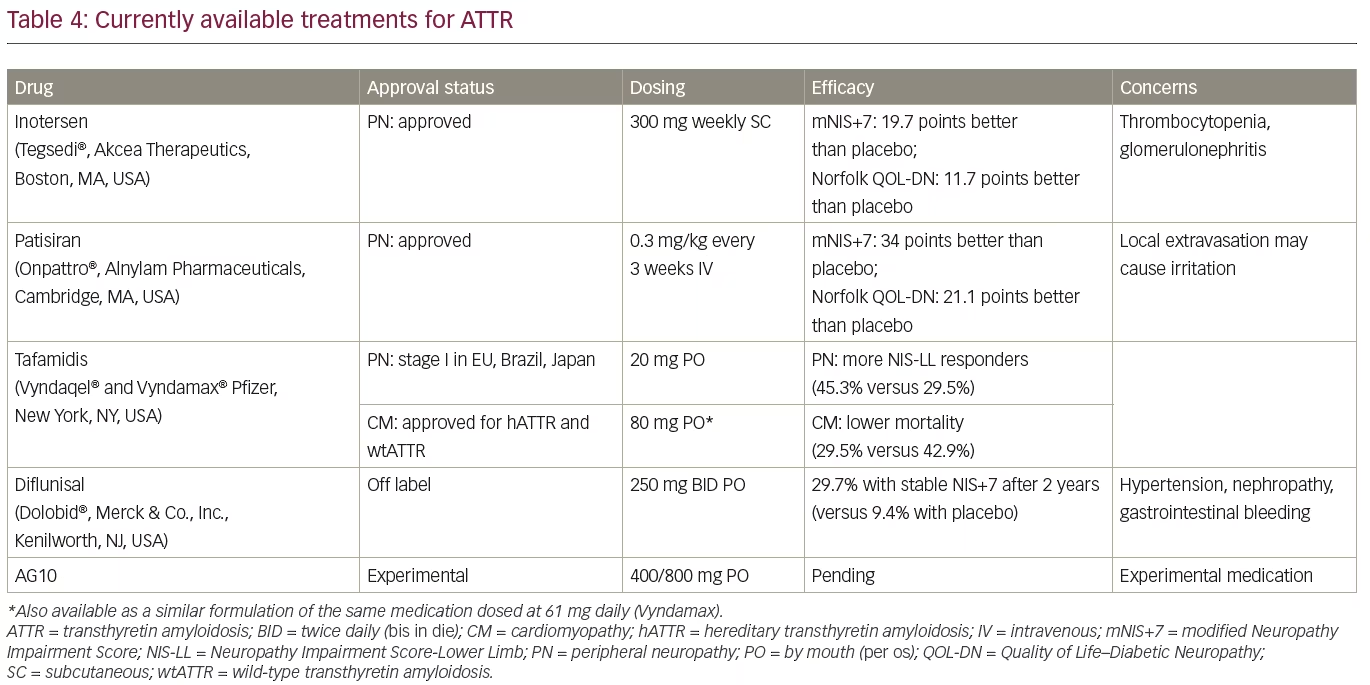
Diflunisal is an NSAID that binds to a thyroxine-binding site on TTR and stabilizes circulating TTR tetramers, leading to reduction on amyloid formation. The use of diflunisal for 2 years in patients with stage I and stage II hATTR-PN reduced the rate of progression and improved patients’ quality of life, whereas 29.7% of patients remained clinically stable compared with 9.4% of patients treated with placebo.84 However, there is a safety concern with the long-term use of NSAIDs (including diflunisal) in patients with significant cardiac disease, renal insufficiency, and the risk of gastrointestinal bleeding.
Overall, while there are no studies allowing head-to-head comparison, TTR knockdown therapies seem to be more effective than TTR stabilizers in the treatment of hATTR-PN.98 Nevertheless, clinical studies demonstrated that TTR stabilizers may delay the progression of neuropathy in ATTR when used early in the disease course. Similar treatment choices are not available for ATTR-CM in clinical practice, and currently only tafamidis is approved for treatment after studies showed a reduction of all-cause mortality and of cardiovascular hospitalizations.68 Studies of knockdown therapy of ATTR-CM are in progress. Tafamidis and diflunisal are oral medications and do not require premedication or laboratory testing.
AG10 is another highly selective TTR stabilizer that is currently undergoing clinical trials. Initial studies showed that it was well tolerated and restored low serum TTR levels to normal, but the final results showing its efficacy from clinical studies are still pending at this time.99 In addition to TTR stabilizers, another option is treatment with amyloid fibril disruptors, including a combination of doxycycline and ursodiol, and limited clinical studies suggest that such a combination may stabilize hATTR-PN.100
Transplantation—liver, heart, domino
Liver transplantation was the first therapy to slow the progression of ATTR, as 95% of circulating TTR in human body is synthesized in the liver, even though the liver in patients with TTR remains otherwise functional. Liver transplantation has been used in the treatment of hATTR since 1990 with more than 2,000 patients receiving a transplant.85 Following liver transplantation, symptoms of hATTR-PN may stabilize or mildly improve in most patients.101,102 However, wild type TTR amyloid may continue to deposit in the heart and peripheral nerves, even after liver transplantation.103,104
Depending on TTR mutation, post-liver transplantation survival at 10 years varies widely from 21–85%.105 Early-onset patients with Val30Met TTR mutation benefit the most from liver transplantation, and poor survival has been noted in most patients with leptomeningeal amyloidosis.85 In a study from Japan, before the availability of knockdown treatments, the estimated probability of survival at 10 years after transplantation for patients with Val30Met TTR mutation was 100% compared with 56% for non-transplant patients.106 Median overall survival of untreated patients with Val30Met TTR mutation and patients who underwent liver transplantation was 11.61 and 24.73 years, respectively.23 In addition to classic liver transplantation from deceased donors, some patients with hATTR underwent domino liver transplantation, whereby the explanted liver allografts from patients with hATTR were transplanted to patients with liver failure. Domino liver transplantation may also lead to development of acquired TTR amyloid polyneuropathy in recipients of domino liver explants, typically at 7–9 years after transplantation.82 Poor nutritional status and longer duration of disease prior to transplantation are also risk factors for poor outcome after liver transplantation.85
Heart transplantation is controversial in patients with hATTR-CM due to the complicated clinical course and continued production of mutant TTR by the native liver.65 Additionally, advanced age and multiple comorbidities will increase the risk of potential complications associated with heart transplantation. Combined liver and heart transplantation may be an option for some patients with refractory heart failure and survival may be greater than with transplanted liver alone.107 The use of knockdown therapies in liver transplant recipients is also being explored (NCT03862807), especially since this should reduce the formation of wild type TTR amyloid fibrils, which may continue after transplantation.
Supportive treatment
hATTR is a multisystemic disorder and requires multidisciplinary approach. Close cooperation between neurologists and cardiologists is essential, and with multisystemic clinical manifestations, other specialties are needed, including nephrologists, gastroenterologists, ophthalmologist, and physical therapists. In addition to treatment of underlying hATTR, most patients with hATTR-PN also require symptomatic therapy of neuropathic pain, dysautonomia, and other associated symptoms, similarly as with other types of peripheral neuropathy. Symptomatic treatment of cardiomyopathy is focused on reducing polypharmacy, maintenance of euvolemia with avoidance of medications that can cause symptomatic hypotension, and consideration of pacemaker placement.65
Conclusion
Overall, new approved treatments of hATTR-PN are changing the natural history of this progressive disease. The currently available treatments mostly slow down and arrest the progression of amyloid deposition, so early diagnosis and timely treatment remain essential for more effective management. Diagnosis of hATTR has been greatly facilitated by non-invasive imaging of cardiac amyloid with nuclear scintigraphy (PYP/DPD/HMDP) and ease of access to genetic testing. Early diagnosis is expected to lead to more effective treatment, especially now that we have several medications showing improved outcomes in treatment of hATTR-neuropathy (inotersen, patisiran, tafamidis, diflunisal) and cardiomyopathy (tafamidis). There may be a role for possible combination of knockdown stabilizers and TTR stabilizers, and reduction of serum TTR levels may also allow lower dosing of TTR stabilizers. Ongoing and future studies will investigate potential benefits of knockdown therapies in hATTR-CM and wtATTR, and new ways to utilize TTR stabilizers in the treatment of ATTR. Future treatments will also target mutant TTR formation in the central nervous system and the eye.