Report based on Presentations at a Satellite Symposium held at the European Paediatric Neurology Society (EPNS) on 25 September 2013 in Brussels, Belgium
At the European Paediatric Neurology Society (EPNS) conference held in Brussels during September 2013, a satellite symposium sponsored by PTC Therapeutics Inc. was held to discuss new perspectives in the management of Duchenne muscular dystrophy (DMD). This symposium was aimed at reviewing the latest evidence concerning the diagnosis and clinical management of DMD, and the development of robust outcome measures for research into this disease.
DMD is a rare X-linked recessive disorder primarily affecting skeletal and cardiac muscle, caused by a mutation in the DMD gene Xp21. It is a devastating progressive disease that is characterised by a predictable clinical evolution. Its incidence is estimated at 1:5,000.1 DMD is predominantly diagnosed at approximately 5 years of age when the patients’ physical abilities differ significantly from those of their peers.2 Patients usually experience loss of ambulation and significant general disability by their early teens, with eventual premature death in their early twenties.3,4
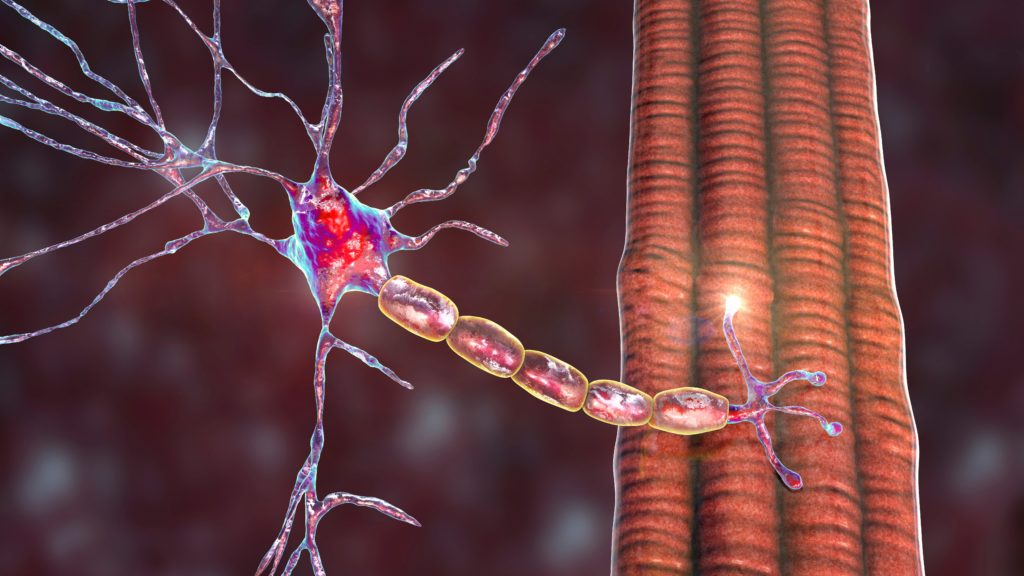
DMD results from the absence of dystrophin, an essential transmembrane muscle protein found in the dystrophin–glycoprotein complex of muscle cells (see Figure 1).5 Dystrophin has a structural role in linking the cortical actin cytoskeleton to the extracellular matrix of muscle fibres via interactions with the dystrophin–glycoprotein complex. It also plays a significant role in cell signalling and in regulating muscle response to oxidative stress.6 The absence of dystrophin inhibits contraction-related conformational changes within muscle tissues, creating disability in motor and other functions.7
No licensed drug treatments for the underlying cause of the disease exist.3 Current therapeutic strategies focus largely on delaying the onset and management of complications. Researchers conducting DMD clinical trials face numerous challenges including the wide variability of disease progression between patients, the need for a standardised treatment approach and difficulties in defining clinically relevant and robust outcome measures.
Current Diagnostic and Management Approaches in Duchenne Muscular Dystrophy
DMD typically follows a pattern of progressive muscle weakness and atrophy, with initial symptoms likely in pre-school aged children. Loss of ambulation may be expected at approximately 10–12 years of age, followed by muscle contractures and progressive scoliosis.3,4 Because of the effect of DMD on the respiratory muscles, respiratory insufficiency is usually exhibited in individuals over 15 years of age, with hypoventilation, especially nocturnal, and insufficient coughing. Progressive dilated cardiomyopathy is also likely above 15 years of age. Life expectancy is heavily dependent on the clinical care provided, which varies significantly between countries and centres. However, natural history studies have demonstrated a progressive shift in the age at loss of ambulation from a mean of 8 to 11 years as a result of improvements in treatment protocols.8,9 (see Figure 2)
The clinical presentation of DMD is primarily proximal muscle weakness, with patients being late to walk, demonstrating a positive Gowers’ sign, and exhibiting difficulties climbing stairs, running and jumping.10 Other signs and symptoms of DMD may include calf muscle hypertrophy, a tendency to walk on the toes, frequent falling and potential developmental delay, including of speech. Incidental laboratory findings of elevated liver enzymes (AST, ALT) are frequently observed in pre-symptomatic boys, leading to further investigation and subsequent DMD diagnosis; a 10–50 fold increase in serum creatine kinase is present from birth in DMD. Gowers’ sign is extremely important in aiding diagnosis at this early stage, and combined with laboratory findings should lead to genetic testing. In exceptional circumstances where genetic tests are negative or unavailable, a muscle biopsy should be taken to confirm the diagnosis.
It is important to consider the specific genetic mutations associated with DMD when diagnosing patients. These mutations are the primary targets for emerging drugs in development to treat DMD but may also be important in assessing a patient’s eligibility for participation in clinical trials. Genetic counselling should be offered to the family of a patient diagnosed with DMD to enable carrier testing and/or pre-natal screening. In previous years, genetic testing centred on multiplex polymerase chain reaction (PCR)11 as it was widely available and inexpensive. However, only some of the genetic deletions or duplications in the dystrophin gene could be detected using this method. The dystrophin gene is the largest gene in the human body, comprising 79 exons12 and genetic diagnosis using PCR can therefore be challenging.
Since the discovery of the dystrophin gene in 1987, technological advances have enabled the detection of DMD deletions from blood samples in approximately 98 % of patients with DMD.13 The majority of laboratories now use multiplex ligation-dependent probe amplification (MLPA) techniques, which evaluate the whole gene to detect deletions and duplications in all 79 exons. It is important to note, however, that a negative MLPA analysis cannot exclude a diagnosis of DMD. If this analysis is negative, genetic sequencing of the complete coding sequence of the DMD gene (Xp21) is necessary to detect point or nonsense mutations that are found to cause approximately 25 % of DMD cases.14 If the clinical presentation is typical of DMD, non-invasive methods are more desirable than invasive procedures. However, if these genetic tests are negative or the clinical presentation is atypical testing should progress to muscle biopsy. A sample that is positive for DMD shows histology of a dystrophic pattern, with active degeneration and regeneration. Immunohistochemistry exhibits an absence of dystrophin staining with three antibodies and only a few ‘revertant’ (dystrophin-positive) fibres. Finally, an immunoblot analysis will show an absence of any dystrophin band.
Following a consensus approach involving a number of patient-centred DMD care groups, recommended care pathways have been published3 that highlight the need for a multi-disciplinary approach due to the complex nature of the disease (see Figure 3). It is important to incorporate physiotherapy and daily stretching, and to utilise assistive technology that can prevent severe contractures otherwise requiring surgical intervention. Psychosocial and educational support should be considered, as should regular cardiac function monitoring with the initiation of angiotensin-converting-enzyme (ACE)-inhibitors once echocardiography is found to be abnormal. Pulmonary assessment and ventilator support become important with disease progression; polysomnography, non-invasive ventilation and cough assist are useful when respiratory problems occur. Glucocorticoids are currently the only clinically available drugs that have shown benefit in DMD, slowing the decline in muscle strength and function.15–21 Corticosteroid treatment, starting in the plateau phase of the disease’s evolution at approximately 4 years of age (when normal childhood growth and development is offsetting the loss of muscle power), should be initiated using a regimen of prednisone 0.75 mg/kg/day or deflazacort 0.9 mg/kg/day.15 In order to investigate optimal corticosteroid regimens in DMD, an ongoing clinical trial Finding the Optimum Regimen for Duchenne Muscular Dystrophy (FOR-DMD) is comparing outcomes in 300 patients receiving regimens of daily prednisone (0.75 mg/kg/day), intermittent prednisone (0.75 mg/kg/day for 10 days on and 10 days off) or daily deflazacort (0.9 mg/kg/day). This study is expected to complete in January 2018.22
As a result of these advances in management of DMD, an increasing number of non-ambulatory DMD patients survives into adulthood. However, many DMD patients do still not receive the recommended care and treatment. This is due to a variety of reasons, including lack of awareness of the latest evidence regarding DMD diagnosis and its management among healthcare professionals, lack of access to reference centres and lack of resources for treatment (CARE-NMD Survey, 2013). There is therefore a need to better communicate the latest recommendation in DMD management.
Several new drug treatments that address the underlying cause of DMD through a variety of means are being investigated. Mechanisms of action of such emerging treatments include over-reading premature stop codons of the DMD gene, exon skipping to restore specific reading frames,23,24 gene therapy and utrophin upregulation.25 The possibility of increasing muscle mass using approaches such as myostatin blockade being investigated.26 Indirect approaches targeting pathophysiological cascade of events downstream of the dystrophin deficiency are under investigation, such as reduction of oxidative stress and improvement of respiratory chain function (Idebenone),27 or targeting the nitrous oxide (NO)-mediated haemodynamic responses to exercise with phosphodiesterase inhibitors.28
What are Meaningful Clinical Outcomes in Ambulatory Duchenne Muscular Dystrophy?
The advent of investigational therapeutic approaches to DMD in the last decade has highlighted the need for robust clinical outcome measures in clinical trials. Optimal outcome measures in DMD need to: be reliable, be able to be validated against other measures, be suitable for multi-centre studies, have known normative ranges, be sensitive to change and to have clinical meaningfulness. Finally, they must also clearly reflect responsiveness to the treatment under study.
Emerging evidence indicates that the 6-minute walking test (6MWT) is valid and reliable as a primary outcome measure in DMD clinical trials. Patients are required to cover as much distance as possible over 6 minutes while walking on a 25 m course on flat ground. The patient may stop if fatigued, but must not sit down, and should resume walking as soon as possible. Distances are recorded at each lap and at every minute. While clearly measuring walking ability, the 6MWT has also demonstrated its viability as a measure of broader physical functioning including endurance.
In the first published natural history study in DMD, the 6MWT was used to characterise ambulation over time in 18 boys with Duchenne or Becker muscular dystrophies and 22 healthy boys, aged 4–12 years, over mean intervals of 58 and 69 weeks, respectively.29 Improvements in 6MWT performance usually occurred in DMD patients by 7–8 years of age, whereas older patients worsened in their 6MWT performance, and older healthy subjects were stable (see Figure 4). The study concluded that the changes in 6MWT performance at 1 year provided a clinically meaningful measure of disease-related limitations on ambulation, and offered a practicable outcome measure for defining DMD natural history and use in clinical trials. Another study evaluated 6MWT data from the placebo arm (n=57) of a phase IIb, international, multi-centre, randomised, double-blind, placebo-controlled, dose-ranging study of the efficacy and safety of ataluren in ambulatory male patients aged ≥5 years with nonsense DMD mutation (nmDMD).30 This study (sponsored by PTC Therapeutics Inc.) was the first placebo-controlled trial in DMD. Results demonstrated that baseline age (≥7 years) alongside baseline 6-minute walking distance (6MWD), as well as performance in selected timed function tests, were strong predictors of ambulatory decline in DMD patients. Because loss of muscle function in DMD is offset by normal childhood growth and development associated with increases in stride length, children aged <7 years may show increases in 6MWD over 1 year despite progressive muscular impairment. The validity of the 6MWT as an outcome measure has subsequently been demonstrated in a number of other clinical studies of DMD, underlining the value of the 6MWD as an outcome measure.31,32
Clinical relevance and clinical meaningfulness are important factors to consider when interpreting changes in 6MWT performance in DMD patients. The phase IIb study of ataluren confirmed that the minimal clinically important difference (MCID) in a 6MWT is 30 m,30 although the meaningfulness of this change depends on the age and stage (baseline walking ability) of DMD in each patient. A persistent ≥10 % decrease in a patient’s walking ability, as measured by 6MWT performance decline from baseline over 1 year, is an important disease marker as this increases the risk of complete loss of ambulation within 4 years.30 It has also been shown that a decline of approximately 30 m from an average performance on the 6MWT to a threshold 6MWD of 325 m is associated with a more precipitous decline in ambulatory function over the subsequent year. Given the limitations of other measures in DMD, the 6MWT has therefore been recommended as the primary outcome measure in ambulatory DMD.30
Another method for validating changes in 6MWT performance in terms of clinical meaningfulness in DMD is to correlate these changes with other measures. The North Star Ambulatory Assessment (NSAA) is a functional ability assessment scale that is useful in DMD patients, with proven reliability and correlation with 6MWT.33–36 Based on an assessment of activities of daily living, such as rising from the floor and standing up from a chair, the NSAA, in combination with the 6MWT, can be effectively used to provide information on different aspects of motor function otherwise unavailable with a single function test.34 When comparing 6MWT performance changes with NSAA changes over 2 years, a linear relationship has been reported, supporting the clinical meaningfulness of both tests.30,37,38 The 6MWT is also strongly correlated with the 10 m run/walk test; a 6-second performance on this test (1.64 m/s) corresponds to 358 m on the 6MWT (correlation coefficient r=0.89; p<0.0001).30,39
In a further study that aimed to correlate 6MWT results with clinically meaningful person-reported outcomes, 1 year changes in the 6MWD and the Paediatric Outcomes Data Collection Instrument (PODCI) scores were reported in 24 boys with DMD and 36 typically developing control boys aged between 4 and 12 years.40 The PODCI is a questionnaire used to quantify health-related quality of life (HRQoL) that is useful in DMD studies.41 Changes in PODCI responses and changes in 6MWD performance were found to correlate strongly (a 30 m change in 6MWD correlates with a clinically meaningful change in QoL, as measured by PODC).40 The degree of perceived ‘meaningful change’ in walking ability, however, differed according to functional capacity. At lower levels of functional ability in DMD, smaller decreases in 6MWD performance resulted in a meaningful change in patient QoL instrument scores, but at higher levels of functional ability, larger performance decreases on 6MWD may be necessary to achieve the same patient QoL score change.40 These factors may affect power calculations and sample sizes needed for clinical trials.
Conclusion
DMD is a debilitating disease for which effective treatment options, addressing the underlying cause of the disease, remain an unmet need. However, significant progress has been made in the knowledge of the natural history of DMD, and management guidelines aim to improve current practice. An effective multi-disciplinary approach is essential. Corticosteroid treatment provides benefits in terms of prolonging ambulation and delaying or preventing the onset of scoliosis. Several treatments in clinical development for DMD are addressing the specific disease-causing genetic mutations, and have highlighted the need for reliable outcome measures in clinical trials. The 6MWT performance correlates well with other established functional outcome and QoL measures. Furthermore, clinically meaningful changes in 6MWT performance have also been defined. However, interpretation of achieved change in 6MWD in DMD may vary by age or functional ability and this should be borne in mind when designing clinical trials. Significant challenges therefore remain in the assessment of upper limb function for both ambulant and non-ambulant boys with DMD and in the development of outcome measures for very young boys with this disease.