Acute hepatic porphyrias are inherited metabolic disorders resulting from a specific enzyme defect in the heme biosynthetic pathway. Acute intermittent porphyria is the most common type and has the most severe presentation.1 Acute episodes can be triggered by surgery, certain drugs, pregnancy, menstruation, infection, or fasting.1
Guillain–Barré syndrome is characterized by rapidly evolving ascending weakness, mild sensory loss and hypo- or areflexia. The nadir of weakness is reached within 2 weeks in 50% of cases and by 4 weeks in 90% of cases.2 In axonal variants of Guillain–Barré syndrome, patients have a more rapid progression of weakness to an earlier nadir, resulting in paralysis and respiratory failure over a few days. Axonal forms of Guillain–Barré syndrome may be similar to acute porphyric neuropathy, in that both have rapidly progressive weakness with respiratory involvement, and autonomic neuropathy.3
This case report describes a patient with acute intermittent porphyria with two separate acute neurologic presentations. Motor neuropathy may be the most objective clinical manifestation that assists in the diagnosis of these uncommon diseases. Acute intermittent porphyria should be considered in patients with recurrent quadriparesis, irrespective of age, gender, racial, and/or ethnic background.
Chart review of the initial hospitalization and follow-up visit revealed a 65-year-old woman of Hispanic ethnicity with poorly controlled type 2 diabetes mellitus and no prior history of weakness, who presented with fever followed by diffuse myalgia, which progressed to flaccid quadriparesis and respiratory failure requiring mechanical ventilation in the setting of underlying infection (cytomegalovirus viremia) within the space of a few weeks. She developed hyponatremia, which upon investigation, was thought to be due to syndrome of inappropriate secretion of antidiuretic hormone (SIADH).
During initial hospitalization, the patient had been treated by the first physician with prednisone and intravenous immunoglobulin for initial presumptive diagnosis of polymyositis, given the markedly elevated creatine kinase (CK; 8,002 U/L), and a thigh magnetic resonance image (MRI) showing abnormal enhancement of the pelvic girdle muscles. Two months later, when she was evaluated by another neurologist in an outpatient setting, the patient had a tracheostomy tube in place and was bedbound. On examination, she was noted to have proximal muscle weakness in the upper extremities (Medical Research Council [MRC] scale for muscle strength of 2/5) and lower extremities (MRC scale 3/5), and distal muscle weakness of upper and lower extremities (MRC scale 3/5) with diffuse hyporeflexia.
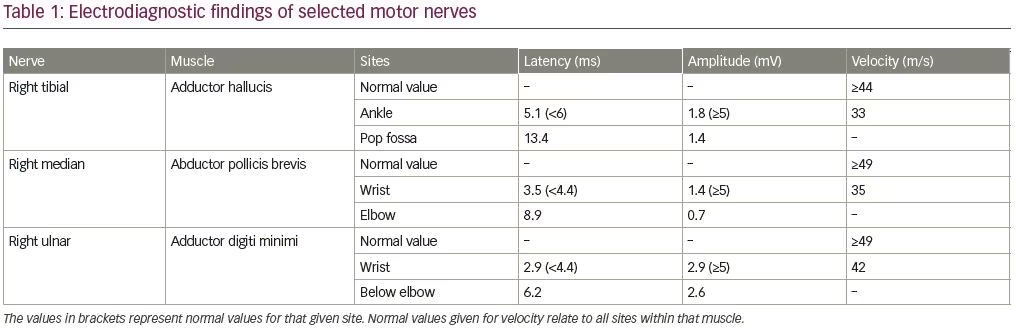
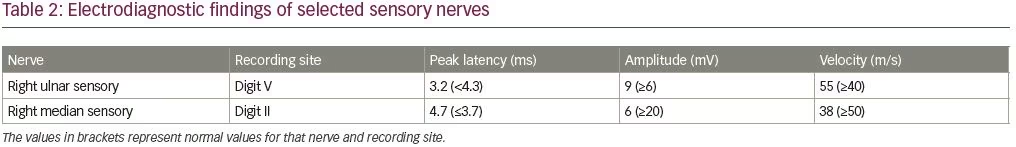
An electrodiagnostic study was interpreted as consistent with diffuse axonal neuropathy with secondary demyelinating changes (Tables 1 and 2). The patient was diagnosed by the neurologist as having an acute motor axonal neuropathy variant of Guillain–Barré syndrome. Axonal loss changes in the sensory nerves were ascribed to underlying diabetes. Sural sensory nerve action potentials were technically difficult to perform due to pedal edema. Needle electromyography examination was performed in the right upper (C5–T1) and lower extremity (L3–S1), and showed 2+ fibrillation potentials with reduced recruitment of normal-looking motor unit action potentials and increased firing rates in all tested muscles.
The patient gradually improved with no further treatment. Within 2 months, she was able to breathe independently and sit unsupported. She had residual proximal muscle weakness (MRC scale 3/5) and distal muscle weakness (MRC scale 4/5) in all four extremities. Five months later, she was seen at the urgent care center because of constipation. She was noted to have a urinary tract infection with a white blood cell count of 19,200/µL, as well as hyponatremia (sodium level 127 mEq/L). Six days later, she developed acute shortness of breath leading to a respiratory arrest requiring intubation. On admission, she was noted to have hemodynamic autonomic instability with quadriplegia and dark tea-colored urine. Pertinent neurologic findings revealed an intact mental status, bifacial weakness, absent gag reflex, and an areflexic quadriplegia with preserved response to pain sensation.
Initial diagnostic work-up including erythrocyte sedimentation rate, C-reactive protein, antinuclear antibody panel, antineutrophil cytoplasmic antibodies, spinal-fluid examination, blood cultures, cytomegalovirus immunoglobulin M, West Nile IgM, and other infectious work-up, including HIV and antibodies (GM1, GD1A, GD1B, and paraneoplastic antibodies) were normal or unrevealing. Cervical spine MRI was unremarkable. CK was mildly elevated at 323 U/L. In the setting of recurrent respiratory failure and acute neuropathy with antecedent infection, porphyria was considered as a potential etiology. Qualitative porphobilinogen in random urine was 6.7 mg/g (normal level [NL] <2.0 mg/g). Further testing revealed elevated urine delta-aminolevulinic acid 36.1 mg/g (NL <5.4 mg/g), uroporphyrin 179.6 µg/g (NL 3.1–18.2 µg/g), coproporphyrin I was 43.3 µg/g (NL 5.6–28.6 µg/g), and coproporphyrin III was 141.6 µg/g (NL 4.1–76.4 µg/g). Erythrocyte porphobilinogen deaminase, fecal fractionated porphyrin, and serum lead were within normal range (NL <10 µg/dL).
The patient received hemin therapy (4 mg/kg/day) for a total of 10 days. Given the occurrence of oliguria and the presence of anasarca, she was started on continuous renal replacement therapy as well. After four doses of hemin therapy, she became unresponsive, with an acute drop in hemoglobin of uncertain etiology and severe hypotension requiring additional vasopressor agents. Continuous renal replacement therapy was stopped and hemin infusion continued for 6 more days. The patient’s mental status and autonomic instability gradually improved, hemoglobin stabilized, the need for vasopressors gradually weaned, and urine output improved; however, her motor neurological status remained unchanged.
In the context of recurrent acute respiratory failure, muscle weakness, and the laboratory findings, the diagnosis of acute intermittent porphyria was made and considered as the most likely type of porphyria. Gene testing, including sequence analysis, however, revealed absence of mutations, deletions, or duplications for the different types of porphyria (aminolevulinate [ALA] dehydratase [ALAD], ALA synthase [ALAS2], coproporphyrinogen oxidase [CPOX], ferrochelatase [FECH], hydroxymethylbilane synthase [HMBS], protoporphyrinogen oxidase [PPOX], uroporphyrinogen decarboxylase [UROD], and uroporphyrinogen synthase [UROS]).
The patient’s hospital course was complicated by infection and other medical issues including severe anemia. She died a few months later.
Discussion
The terms “porphyrin” and “porphyria” are derived from the Greek word porphyrus, meaning purple. Acute intermittent porphyria affects people of all races and regions, but it is more common in people of Northern European descent.4 Clinical illness usually develops in women in their teens or 20s, but onset can begin after menopause in rare cases.5 Our patient, however, was of Hispanic descent, postmenopausal, without a previous diagnosis of porphyria, and no known family members had this disorder.
Porphyria refers to rare metabolic disorders with enzymatic defects in the biosynthesis of heme.1 The rate of heme synthesis depends on the expression of the erythroid-specific ALAS gene. In the liver, ALAS1 has a key regulatory role in heme synthesis, and is rate-limiting for hepatic heme synthesis.6–8 A regulatory heme pool controls the expression of the hepatic ALAS1 gene and the transport of ALAS1 into mitochondria. Acute porphyria attacks are associated with increased production of heme precursors and can be triggered by severe fasting or dieting, alcohol, certain drugs (especially barbiturates, hydantoins, rifampin, sulfonamides, and endogenous steroid hormones [estrogen and progesterone]) and intercurrent illnesses or stress.
The clinical features are similar during acute porphyric attack in all acute porphyrias. However, the attacks are less frequent and less severe in variegate and hereditary coproporphyria.9 Most of the clinical manifestations are due to the effects of precursors of heme on the nervous system. Colicky abdominal pain is the most common presenting symptom, usually affecting the lower abdomen and lasting from several hours to days. It is gradual in onset and escalates in severity. Many patients have red to brown urine that may darken when exposed to air, light, and warmth.
Generalized weakness may sometimes progress rapidly to quadriparesis and acute respiratory insufficiency. Hyponatremia is a common electrolyte abnormality that occurs during acute attacks. Factors that contribute to hyponatremia include SIADH, vomiting, and resuscitation with high volumes of dextrose solutions given intravenously.10 Our patient had dark tea-colored urine, recurrent respiratory failure, quadriplegia, and hyponatremia in the setting of active infection.
The most common presenting finding in patients with acute intermittent porphyria is severe abdominal pain. Other gastrointestinal symptoms include nausea, vomiting, diarrhea, or constipation.11 Our patient did present with constipation in the setting of urinary tract infection. Common neurologic manifestations of acute porphyrias include severe pain, peripheral neuropathy, muscle weakness, difficulty swallowing and other bulbar symptoms, confusion, delirium, and seizures.11,12
Porphyric neuropathy affects both the autonomic and somatic peripheral nervous system. Acute intermittent porphyria is the most common type of hepatic porphyria associated with neuropathy. Neuropathy occurs in 20–68% of patients with porphyria; the typical pattern of porphyric neuropathy is predominantly motor axonal neuropathy.13–15 Motor neuropathy is normally symmetrical and begins in the proximal part of the upper extremities; however, the patterns of motor involvement are variable.16,17 About 50% of patients experience onset in the arms with proximal distribution. Our patient had proximal weakness during her first attack and dysautonomia after her second one. Axonal motor neuropathy is the characteristic electrophysiologic pattern of acute porphyric neuropathy,12,18 and usually develops 3–75 days after the initial symptoms and progresses to maximum deficit within 1 month.
The pathogenesis of porphyric neuropathy is complex, but overproduction of ALA via direct neurotoxicity, oxidative damage, and modification of glutamatergic release may initiate the neuronal damage.19 The electrodiagnostic study of our patient after the first hospitalization revealed axonal loss sensorimotor polyneuropathy with secondary demyelinating changes. The sensory axonal loss changes were ascribed to underlying diabetes. The motor axonal changes were attributed to acute motor axonal neuropathy. In retrospect, in the context of recurrent attacks, tea-colored urine, dysautonomia, SIADH, and lab findings, acute motor axonal neuropathy diagnosis was thought to be unlikely.
Given the markedly elevated CK (8,002 U/L) and a thigh MRI showing abnormal enhancement of the pelvic girdle muscles during first hospitalization, the patient had been treated with corticosteroids and intravenous immunoglobulin to target both the differential diagnosis of polymyositis and Guillain–Barré syndrome. Later, the elevated CK was ascribed due to rhabdomyolysis. Rhabdomyolysis has been reported in the acute phase of porphyria and porphyria, and this should be considered in patients who present with abdominal pain and rhabdomyolysis.20
The clinical presentations of acute porphyrias are nonspecific. Hence, laboratory evaluation is vital for the diagnosis of acute porphyria. Qualitative or semi-quantitative urinary porphobilinogen, performed on a single random urine sample, is the most important rapid test for the diagnosis of acute porphyria.21 Elevation of porphobilinogen, and delta-ALA are the most frequently noted abnormalities in acute intermittent porphyria. Porphobilinogen in urine may be converted non-enzymatically to uroporphyrin, and hence, although the defect in acute intermittent porphyria lies in hepatic porphobilinogen deaminase, there may be increased urinary uro- and copro-porphyrin levels. Our patient had elevated porphobilinogen, delta-ALA, uro- and copro-porphyrin levels. Differential diagnosis includes lead neuropathy as it is associated with a defect in the heme metabolic pathway and increased urinary delta-ALA. Our patient had elevated delta-ALA with a normal serum lead level.
DNA analysis is the most accurate way to screen symptom-free patients to facilitate correct treatment and proper genetic counseling of family members who are at risk.22 Molecular heterogeneity limits the potential of DNA diagnostics in acute intermittent porphyria, and the value of DNA analysis is reduced in cases in which a mutation is unknown. A molecular and biochemical study of 196 patients with known acute intermittent porphyria showed that, in two cases, long polymerase chain reaction (PCR) fragments and sequencing of all exons, exon-intron boundaries, and 5′- and 3′-untranslated regions did not reveal a genetic defect.23 Another study in individuals with a proven biochemical diagnosis of acute intermittent porphyria, reported that of 265 individuals, 255 had a variant detected by sequence analysis, five had a variant detected by gene-targeted deletion/duplication analysis, and in five no variant was detected.24
In the context of her clinical presentation, recurrent acute attacks, tea-colored urine and dysautonomia, SIADH, elevated levels of urinary porphobilinogen, delta-aminolevulinic acid, and uroporphyrin, acute intermittent porphyria was considered. Although a mutation could not be identified in this patient, acute intermittent porphyria could not be excluded given her clinical profile presentation and the noted biochemical findings. In fact, identification of the mutation is not a requirement to make the diagnosis. Similar to our case, diagnosis can be made based on clinical history and biochemical abnormalities.
Initial management of acute porphyria also includes a search for, and avoidance of, precipitants, and discontinuation of all potentially harmful drugs. In moderate-to-severe attacks of acute porphyria, intravenous heme is the treatment of choice. ALA excess may be associated with a cytotoxic effect on neurons, which could be diminished by hemin infusion.25 Typically, about 4–5 days of treatment with heme, 3 mg/kg bodyweight/day, is required to resolve symptoms following an acute attack. Our patient received hemin infusion for a total of 10 days. Side effects of hemin therapy include obliterative thrombophlebitis, coagulopathy, tachyphylaxis, and iron overload, when used frequently or for long periods (>3 years). As hemin degrades rapidly and causes infusion-site phlebitis and coagulopathy, reconstitution with human albumin is recommended.26
A breakthrough in the treatment of acute hepatic porphyrias became recently available and would change the approach to treatment. Givosiran, a novel ALA synthase 1 (ALAS1)-directed small interfering RNA (siRNA), was approved by the US Food and Drug Administration in November 2019. Givosiran covalently links to a ligand to enable specific delivery of the siRNA to hepatocytes. This results in down-regulation of ALAS1 mRNA and prevents accumulation of neurotoxic delta-aminolevulinic acid and porphobilinogen levels.27 Givosiran was shown to significantly reduce the rate of porphyria attacks that required hospitalizations, urgent healthcare visits, and intravenous hemin administration at home compared with placebo.28
A phase I clinical trial to determine the safety and efficacy of givosiran in patients with a type of acute hepatic porphyria, known as acute intermittent porphyria, was completed in 2017 (ClinicalTrials.gov Identifier: NCT02452372). The study also showed that givosiran reduced the need to use hemin by 48% (hemin is usually administered in the event of an acute porphyria attack). Patients from the trial were given the opportunity to take part in a phase I/II open-label extension trial on the long-term safety and efficacy of givosiran. The use of hemin was also reduced by up to 88% in treated patients compared with those on placebo.29,30
Discontinuation of triggering factors and symptomatic treatment can improve prognosis for recovery and significantly reduce mortality. Neuropathy prognosis depends on the extent of axonal degeneration, which can be irreversible and would result in partial recovery at best. Recovery usually occurs over many months, and after repeated attacks, cumulative deficits may cause permanent weakness. Early recognition and the importance of obtaining a thorough history are crucial for avoidance of diagnosis of mimickers, as happened in our case. Misdiagnosis would lead to erroneous therapy and delay institution of appropriate therapy. With the advent of genetic therapy, we are hopeful that a new paradigm will emerge in the diagnosis and treatment of acute hepatic porphyria.