
Spinal muscular atrophy (SMA) is a progressive neuromuscular disorder affecting approximately 1 in 10,000 births. It is characterized by predominantly proximal muscle weakness as a result of degeneration of anterior horn cells of the spinal cord.1 In most patients with SMA, the disease is caused by a homozygous deletion or mutation of the telomeric survival motor neuron gene (SMN1) on chromosome 5q13. 2,3 SMN1 produces the SMN protein. This intracellular protein is found in many tissues, and in particular, is expressed in high levels in spinal motor neurons.4
Centromeric SMN2 is a paralogous gene that differs from SMN1 by only five nucleotides.5 Only one of these nucleotides is present in the coding region of exon 7, but it results in a cytosine-to-thymine substitution that causes the absence of exon 7 from SMN2 during transcription.3 The result is a truncated, unstable protein that is rapidly degraded. However, the exclusion of exon 7 from SMN2 is incomplete, so about 10% of the SMN2 transcripts are full-length and produce a small number of functional proteins.5 In SMA, absence of SMN1 results in relative deficiency of the SMN protein, and this is thought to result in the clinical presentation. Although the SMN protein is known to be involved in several cellular functions, it is not yet understood why its relative absence results in the SMA clinical phenotype.6
The variability of phenotype is striking, particularly given that most patients with SMA have the same genetic defect. Three major SMA types were defined at the International Consortium on Spinal Muscular Atrophy in 1991, but there have since been modifications to the categorization scheme and five types are now recognized.3 SMA phenotypes are classified based on age of onset and maximum motor function achieved. They range from type 0 in which neonates present with severe weakness and hypotonia at birth and have a history of decreased fetal movements in utero, to type 4 in which adults develop progressive weakness but generally retain the ability to ambulate for most of their life.
Typically, milder phenotypes of SMA have been associated with higher number of SMN2 copies because more copies of SMN2 can produce more full-length transcripts to compensate for the loss of SMN1. This appears to be the case for about 95% of patients with SMA who have a homozygous absence of SMN1. However, in the approximately 5% of patients who do not have complete absence of SMN1, SMN2 copy number does not necessarily seem to modulate phenotype.5 In these patients, there is a deletion or gene conversion of one copy of SMN1 and a subtle mutation on the other chromosome, resulting in compound heterozygosity. These patients can be more difficult to diagnose because they do not lack both copies of SMN1, as is typically seen in SMA.
Once a diagnosis of SMA is clinically suspected, the current method of confirmation is by molecular genetic sequencing in order to identify a homozygous absence of SMN1. While the next-generation sequencing (NGS) that is now commercially available has improved our ability to detect mutations consistent with SMA compared to earlier technologies utilizing polymerase chain reaction (PCR), current genetic testing is still limited.7 While the testing can identify larger or previously described mutations, it may not be able to detect certain variants such as inversions, gene conversion events, translocations, short tandem repeats, or segmental duplications.8 Additionally, according to one of the companies that performs commercial genetic testing, “sequence changes in the promotor, non-coding exons, and other non-coding regions are not covered” by their assay testing for SMA.8
With novel therapeutics on the horizon, accurate diagnosis is of the utmost importance. Nusinersen is an antisense oligonucleotide approved by the US Food and Drug Administration (FDA) in December 2016 that alters the splicing of the SMN2 mRNA transcript, allowing for increased production of full-length SMN protein, and has been shown to improve developmental and motor milestones. In addition, gene therapy clinical trials have recently concluded with remarkably positive results in SMA type 1, and are being conducted in SMA type 2 and 3. By using adeno-associated viral vectors (AVXS-101) to introduce a functional copy of the SMN gene to motor neurons, they can achieve production of normal amounts of the SMN protein and improve motor neuron function.9,10 In fact, this proprietary gene therapy was recently approved by the FDA on May 24, 2019 for use in patients <2 years old with SMA type 1.
While the technology to accurately diagnose genetic conditions has improved, it remains a challenge to diagnose compound heterozygotes due to the near-identical structure between the SMN1 and SMN2 genes. It remains our duty to describe as many genetic mutations as possible that result in SMA phenotypes so we can continue to advance our diagnostic capabilities and open up treatment opportunities to our patients. We will now present two cases illustrating patients with milder SMA phenotypes who were found to be compound heterozygotes with novel sequence variants.
Case presentations
Patient 1
Our first patient is a 28-year-old female. Unfortunately, she was a poor historian, so some details of her history were equivocal. She reportedly walked at 1 year of age but developed weakness and later gradually lost the ability to ambulate. She was diagnosed with SMA at age 9 at a different hospital and we are unsure how she was diagnosed at the time. She began exclusively using a wheelchair around age 22. There was no family history of SMA; however, this aspect of her history was also unclear. She was estranged from her mother since childhood and her father died in a car accident at age 41. She has six unaffected siblings, but we were unable to determine if they were all from the same parents.
Her exam was notable for a prominent scoliotic deformity. She had full extraocular mobility, but had weakness of bilateral orbicularis oculi muscles. Her speech was hypophonic and nasal, and she had prominent tongue atrophy and fasciculations. She had diffusely low tone, but also had several joint contractures. She had marked, largely symmetric weakness of all extremities except for more pronounced distal weakness in her left lower extremity, with associated diffuse muscle atrophy and decreased muscle bulk. Her deep tendon reflexes were absent.
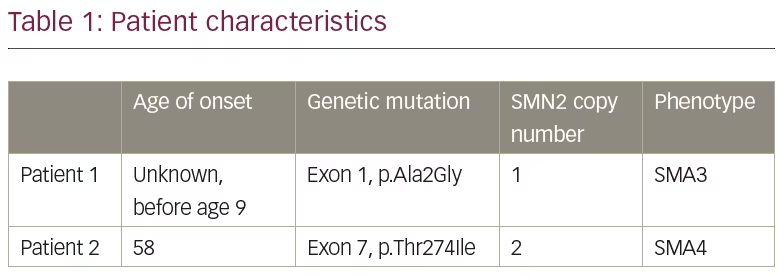
Genetic testing on a specimen of the patient’s saliva was performed by Invitae Laboratories (San Francisco, CA, USA). The Invitae Spinal Muscular Atrophy Panel was used which employs NGS technology to perform full-gene sequencing and deletion/duplication analysis. This genetic testing identified one pathogenic variant in SMN1. Furthermore, an indeterminate variant was identified on exon 1, in which alanine was replaced with glycine at codon 2 (p.Ala2Gly), as indicated in Table 1. Invitae explains that their assay cannot determine whether a sequence variant outside of exon 8 (typically referred to as exon 7) is located on SMN1 or SMN2 due to the nearly identical structure between the two genes.8 She was found to have one copy of the SMN2 gene.
Patient 2
Our second patient is a 67-year-old female who had difficulty running and frequent falls in childhood. Around age 58, she developed more noticeable slowly progressive extremity (lower more than upper) weakness and atrophy that was evidenced by difficulty raising out of a chair and walking upstairs. She has a brother who is 60 years old with SMA type 3 who became symptomatic at age 13 and started using a wheelchair around age 53. She has two other brothers who are unaffected and a 34-year-old healthy child. Her mother died at age 80 from congestive heart failure and her father died at age 93 with multiple medical conditions.
Her exam was notable for intact cranial nerves including normal orbicularis oculi strength and absence of tongue atrophy or fasciculations. She had bilateral, slightly asymmetric weakness of her triceps, wrist extensors, finger flexors, and thumb and 5th digit abduction. In her lower extremities, she had moderate proximal and distal weakness. She had absent deep tendon reflexes in her triceps and lower extremities bilaterally, with 1+ reflexes in her biceps and 2+ reflexes in her brachioradialis muscles. She was able to walk with high steppage gait bilaterally, but was unable to walk on her heels or toes. She also had high-arched feet and hammertoes.
Genetic testing on a specimen of the patient’s saliva was performed by Invitae Laboratories and identified one pathogenic variant in SMN1. She also had an indeterminate variant identified on exon 7, in which threonine is replaced with isoleucine at codon 274 (p.Thr274Ile), as indicated in Table 1. Again, given the sequence similarity between the SMN1 and SMN2 genes, the lab was unable to determine if this sequence mutation was on the SMN1 or SMN2 gene. The patient was noted to have two copies of SMN2.
Family testing was done of the patient’s affected brother and revealed that he also had one pathogenic variant in SMN1, as well as the same indeterminate variant on exon 7. Like our patient, he also had two copies of SMN2. Unfortunately, our patient was subsequently lost to follow-up and we were not able to obtain additional information about her brother’s symptoms.
Discussion
The similarity between these patients was that neither had the typical homozygous deletion of SMN1. They were both found to have only a single deletion of SMN1 and an indeterminate sequence variant. One of the challenges we faced in interpreting these results was that the sequencing that is commercially available cannot distinguish between SMN1 and SMN2 due to their nearly identical structures. Therefore, the genetics lab could not definitively tell us which gene contained the sequence variant. As a result, they could not determine whether or not the sequence variant was pathogenic.
If the variant is on their single remaining copy of SMN1, it would likely mean that the variant is pathogenic or, in other words, the cause of SMA. If the variant is on a copy of SMN2, the variant is likely silent and does not contribute to their disease. Given that both of their phenotypes were consistent with SMA, and given that patient 2’s brother had an identical mutation to our patient, and had progressive weakness with onset in his teenage years, we concluded that the sequence variant was likely on the remaining copy of SMN1. By definition, these patients are compound heterozygotes.
Both of our patients had two or fewer copies of SMN2. By conventional standards, they should be classified as SMA type 1. However, both patients have much milder phenotypes consistent with SMA type 3 and type 4, respectively. Interestingly, patient 2’s brother has an identical mutation as our patient, yet his presentation was more severe with symptom onset at age 13. In our compound heterozygous patients, SMN2 copy number does not necessarily seem to modulate phenotype. Rather than SMN copy number contributing to SMA phenotype, perhaps the specific mutation on the remaining SMN1 gene matters more in terms of clinical phenotype.
Yamamoto et al. also found that this may be the case. They described six SMA patients with compound heterozygous mutations. Three of these patients had the same missense mutation on exon 1 in which alanine is replaced with valine at codon 2. This mutation is very similar to the missense mutation identified in patient 1 (exon 1, alanine replaced with glycine at codon 2). All three of the patients they described carried one copy of SMN2, and two of the three patients had a milder phenotype consistent with SMA type 3.5
Patient 2 was found to have a cytosine-to-thymine mutation on exon 7. As described earlier, the difference between SMN1 and SMN2 is five nucleotides, but a single one is present at position 6 of the coding region of exon 7.5 Although our patient’s cytosine-to-thymine mutation on exon 7 is in a different position, perhaps the mutation resulted in less full-length SMN1 transcripts from being produced, similar to how the mutation on SMN2 results in the production of less full-length transcripts.
As indicated previously, there are limitations to the current technology used commercially for genetic testing. In addition to challenges distinguishing between the two genes, the testing that is used is focused solely on particular exons of interest and may miss mutations in non-coding regions that may somehow modulate genetic expression in ways not yet understood. This may be another reason for, or contribute in some way to, the wide spectrum of phenotype seen in patients with SMA.
Conclusion
We described two patients with previously undocumented sequence variants resulting in SMA compound heterozygotes. While the genetic testing could not yet distinguish which gene contained the indeterminate variant, we believe that the mutation is on the remaining copy of SMN1, resulting in pathogenicity. Both patients have mild phenotypes relative to how few copies of SMN2 they have. This supports the idea that intragenic mutations in SMN1 may correlate more with severity of phenotype rather than copy number of SMN2.1,5
At this time, it remains a challenge to diagnose compound heterozygotes due to the near-identical structure between SMN1 and SMN2. As genetic testing methods continue to improve, we may be able to better correlate specific mutations with disease severity. In fact, current technologies are being studied that can permit measurement of the amount of functional SMN protein in a patient’s serum.6 Not only can this be correlated with disease severity and give us a new way of defining the different subtypes of SMA, it may also be used to objectively monitor improvement in patients being treated with gene-therapy by observing increases in the SMN protein in the patient’s serum.6
We must continue to acquire as much genetic information as possible, as it will ultimately help expand our understanding of the various mutations that can lead to disease states. A future direction may be to perform whole-exome or even whole-genome sequencing, both to help with diagnosis of the more complex genetic cases, and to provide us with an opportunity to learn more about genetic diseases.











