Chronic inflammatory demyelinating polyneuropathy (CIDP) is a rare but challenging neuroimmunologic peripheral nerve disease with a chronic progressive or relapsing-remitting course.1,2 Effective maintenance therapies are critical for managing the condition, maintaining a response, and preventing relapse.3 In CIDP, symptoms result from demyelination of the peripheral nerves.4 The etiology of the disease is as yet unknown but the pathology is associated with neuroinflammation which may be responsive to immunomodulating agents, suggesting an autoimmune cause.5
There are different forms of CIDP. The classic and most common form of CIDP manifests as a loss of strength (usually in proximal and distal limb muscles) and sensation (usually symmetrical and distal). This results in weakness, loss of dexterity and grip strength, as well as gait disturbance and imbalance.6,7 The disease occurs most commonly in individuals of 40–60 years of age and has a progressive or relapsing-remitting course. Occasionally, the disease can have a monophasic course. The frequency of these forms was investigated in an epidemiological study (n=165) which found the disease was relapsing-remitting in 25.8%, chronic progressive in 61.9% and monophasic in 12.3% of patients.8 The epidemiology of CIDP varies widely in different reports.9,10 This may be a result of variable awareness of the condition and diagnostic approaches at different treatment centers. The estimated incidence of CIDP is 0.5–1.6/100,000/year with a prevalence of 0.7–9.0/100,0009–11 (equating to an approximate range of 3,250–29,000 people in the USA).
Patients with CIDP bear a severe burden in terms of fatigue, pain, anxiety, depression and disability; the condition also places a burden on family members and caregivers.12,13 These and other factors substantially contribute to a poorer quality of life for many patients. The aim of CIDP treatment is to reduce symptoms, improve functional abilities, prevent relapse and maintain long-term remission. Various maintenance therapies can be used to treat CIDP. The oldest are corticosteroids, which decrease inflammation and inhibit the immune system; they remain an important part of the standard of care for long-term treatment of this disease.14,15 These are effective and have a low cost. Recent data indicate that corticosteroids can achieve long therapy-free remission in many patients.14,16,17 The use of pulsed corticosteroid treatment can provide efficacy and decrease the side effects associated with long-term use.16
Another widely used class of treatment is immunoglobulins, whose mode of action in CIDP is largely unknown. These are believed to have multiple immunomodulatory effects such as interfering with the complement system and possibly competing with nerve-targeting autoantibodies that may be present in this disease.18–20 These are effective and are perceived to cause fewer side effects than corticosteroids in long-term use; however, there have been no head-to-head studies to support this.19 Many immunoglobulins, however, have limitations including the need for frequent intravenous (IV) dosing, which is inconvenient and requires administration from healthcare professionals resulting in additional cost. Current guidelines recommend that IV immunoglobulin (IVIg) should be used as a first-line treatment option in CIDP.15,21
Other less frequently used alternatives for CIDP treatment include: cyclophosphamide,22 rituximab,23 plasmapheresis,24,25 and autologous stem cell transplantation.4,26 These approaches are usually reserved for treatment-resistant CIDP. Clinical study evidence supporting them comes from small studies and more extensive evaluation is needed to determine their real benefits. In addition, immunomodulatory agents such as cyclophosphamide have various safety issues,22 while stem cell treatment requires considerable medical resources, and thus are unsuitable for general use in CIDP.
Unmet needs in chronic inflammatory demyelinating polyneuropathy treatment
There are a variety of unmet needs in the effective management of CIDP and these are summarized in Table 1.27–31 Despite the range of treatments available for CIDP, there is a need for effective therapies that are less burdensome and have fewer side effects compared with those currently available. Treatment with IVIg is commonly used and has demonstrated clinical efficacy over many years, but it has its drawbacks.7,32–34 In particular, this treatment has a relatively high risk of various systemic side effects, notably, rash, headache, nausea and diarrhea, some of which can last over an extended period of time.35 In addition, all immunoglobulin products, including subcutaneous formulations, have a risk of serious thromboembolic events.36,37 These may limit the tolerability of IVIg and necessitate the use of alternative treatments.

A further drawback is that IVIg requires repeated venous access;38 this can become difficult in patients who have received long-term therapy and patients with limited venous access who potentially need port placement. In addition, patients may require more frequent infusions to manage their disease.38–41 Patients may also have issues with comorbidities and other factors such as hypertension, blood volume management, and thrombosis.42–47 The logistics of IV infusions are also limitations; patients either need to travel to a treatment center or a nurse needs to visit them at home on a regular basis.7,48 The infusion normally takes 4–6 hours but duration can be longer depending on infusion rates and volumes as tolerated, which often interrupts work, parental and social activities.7
An alternative that addresses some of the issues arising from IVIg treatment is subcutaneous immunoglobulin (SCIg; Hizentra® [CSL Behring, King of Prussia, Pennsylvania, USA], immune globulin subcutaneous [human] 20% liquid).49–52 This option has been approved for use as maintenance therapy in adults with CIDP53,54 and has similar efficacy to IVIg.51,55–57 Clinical studies show SCIg is associated with fewer systemic side effects, and for many patients may be a more convenient alternative to IVIg for maintenance treatment in CIDP.
Intravenous and subcutaneous immunoglobulin therapies in chronic inflammatory demyelinating polyneuropathy
The features of SCIg compared with IVIg administration are summarized in Table 2. The advantages of SCIg over IVIg include:
- No need for IV access
- A smaller volume for infusion based on equivalent dose in grams
- SCIg can be self-administered and the infusion time is shorter (in some cases less than a quarter of the time) providing convenience for the patient.33,50–52
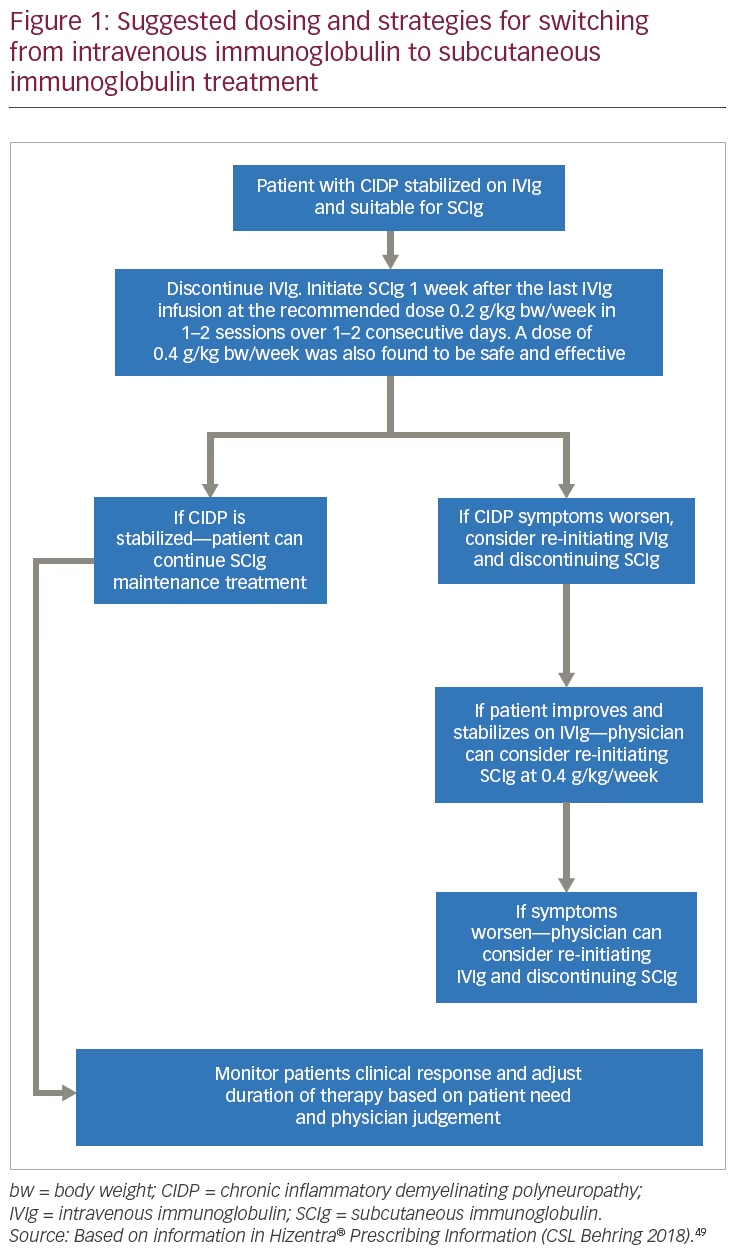
In CIDP, the choice of IVIg or SCIg may be based on patient-specific factors such as occurrence of IVIg-related systemic adverse reactions, ease of venous access, comorbidities, need for greater autonomy, and inability to travel to a distant treatment center.33,51,52 The characteristics of patients with CIDP who can be considered appropriate for switching to SCIg are summarized in Table 3 and the suggested dosing/treatment strategy is given in Figure 1. Patients must have responded to, and be stabilized on IVIg therapy, before being considered for switching to SCIg.49 When giving SCIg in CIDP, infusions are administered into sites on the abdomen, thigh, upper arm, or side of upper leg/hip using a different site for each infusion.49


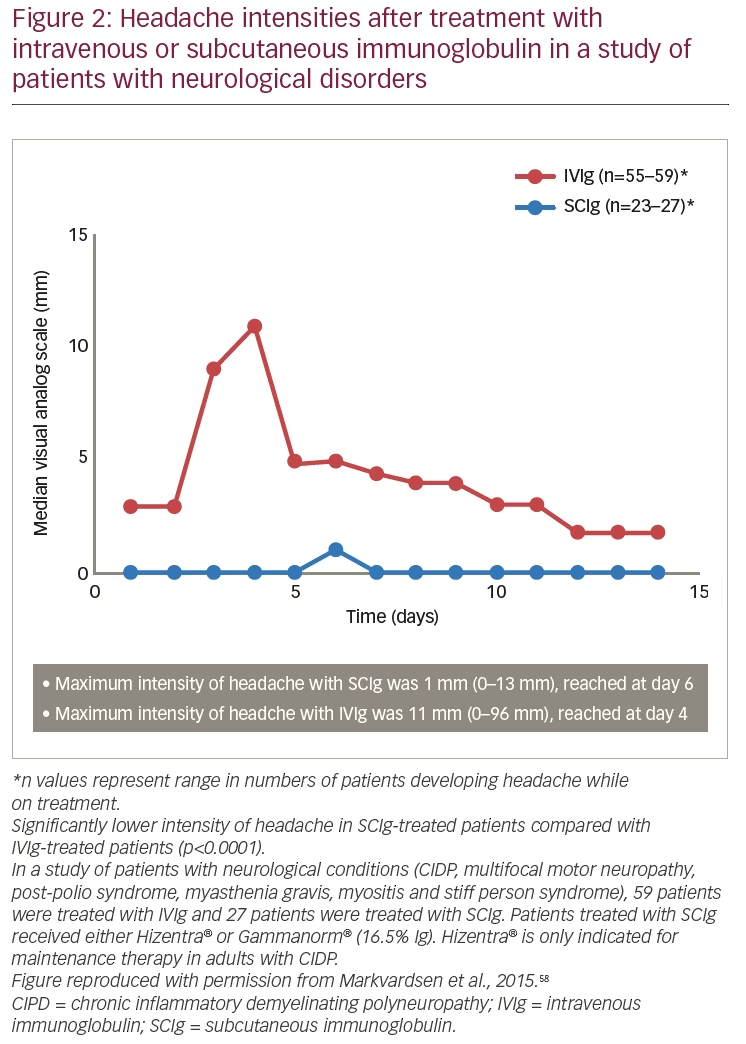
Systemic adverse reactions are possible with any immunoglobulin therapy, including SCIg; however, there is evidence that the rate of systemic adverse reactions is lower with subcutaneous infusions. Systemic adverse reactions known to occur with IVIg include flushing, fever, muscle aches, tiredness, headache, dizziness, neutropenia, weakness, chest pain, tachycardia, blood pressure changes, aseptic meningitis, thrombosis, and renal failure.33,34 A study of patients with neurological diseases (n=86, including patients with CIDP) found a lower frequency and intensity of headache for patients treated with SCIg (n=27, day 6 visual analogue scale: 1 mm) compared with those treated with IVIg (n=59, day 4 visual analogue scale: 11 mm) (p<0.0001) (Figure 2). A significant advantage was also seen for SCIg over IVIg for nausea (p<0.0001).58 Although nausea is a less common event with SCIg than with IVIg, it can continue for several days after administration. Overall, it has been shown that SCIg may result in fewer systemic adverse reactions.34,51,59
It should be noted that thrombosis may occur with immunoglobulin products, including SCIg. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheters, hyperviscosity, and cardiovascular risk factors. For patients at risk of thrombosis, it is necessary to administer SCIg at the minimum dose and infusion rate practicable. It is also necessary to ensure adequate hydration in patients before administration and to monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity.60
The Polyneuropathy and Treatment with Hizentra® (PATH) trial—a pivotal study of subcutaneous immunoglobulin in chronic inflammatory demyelinating polyneuropathy
Key evidence supporting SCIg use in the treatment of CIDP has come from the pivotal phase III, Polyneuropathy and Treatment with Hizentra® (PATH) trial.52,61 This is the largest study of CIDP therapy to date, recruiting, at treatment centers worldwide, a total of 172 patients who were ≥18 years old and had definite or probable diagnosis of CIDP according to the European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) 2010 criteria.21 All patients in the study were required to be dependent on IVIg treatment (i.e., relapsed after stopping treatment and then re-stabilized on IVIg). Participants were stabilized on IVIg during a pre-randomization phase and were then randomized 1:1:1 to 0.2 g/kg body weight (bw) (low-dose), or 0.4 g/kg bw (high-dose) SCIg, or placebo weekly for 24 weeks. The administration of SCIg involved infusion at up to eight infusion sites (2–8) with an average volume of 20 mL (maximum 50 mL) per site with a maximum total volume of 140 mL/session. The median infusion time was approximately 1 hour.49,52
The patient demographics were similar in the three treatment groups. For the 0.2 g/kg bw, 0.4 g/kg bw groups and placebo, the proportions of male patients were 74%, 53%, and 65%, respectively; mean ages were 58.9, 55.2, and 57.6 years, respectively; and mean disease durations were 2.8, 3.3, and 2.7, years, respectively.52
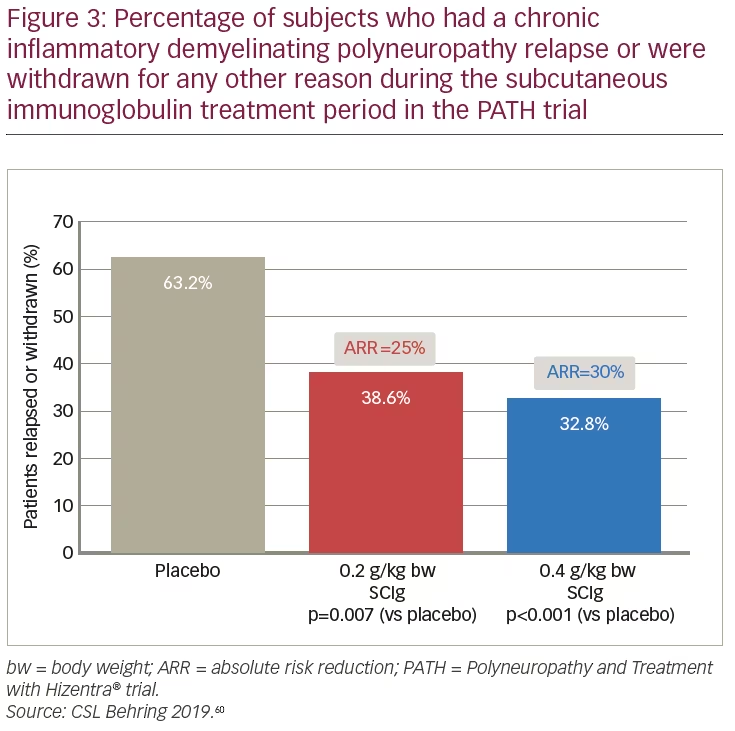
The results of the PATH study demonstrated the efficacy of both SCIg doses for the maintenance of CIDP. Both the 0.2 g/kg bw and 0.4 g/kg bw doses showed superiority over placebo for the primary endpoint: the percentage of patients who relapsed or withdrew from the study (38.6%, 32.8%, and 63.2%, respectively) (Figure 3).61 Absolute risk reduction for the primary endpoint was 25% (p=0.007) and 30% (p=0.001) for the 0.2 g/kg bw and 0.4 g/kg bw doses of SCIg versus placebo, respectively (Figure 3). The relapse sensitivity analysis showed that treatment with SCIg prevented relapse in higher proportions of patients than placebo: for 0.2 g/kg bw, 0.4 g/kg bw, and placebo, rates of patients without relapse were 67%, 81%, and 44%, respectively; absolute risk reduction was 23% (p=0.01) for 0.2 g/kg bw and 37% (p<0.0001) for 0.4 g/kg bw SCIg.52
Among secondary endpoints of the PATH study, SCIg low- and high-dose treatments maintained functional status in various measures compared with the placebo group, for whom these worsened. These parameters included: Inflammatory Neuropathy Rasch-built Overall Disability Scale (I-RODS) (centile score) (overall p=0.0002); dominant and non-dominant grip strength (overall p=0.0223 and p=0.0026, respectively); Inflammatory Neuropathy Cause and Treatment total score (INCAT) (overall p<0.0001) and the Medical Research Council (MRC) sum score (overall p=0.0026).52 An exploratory endpoint investigated ease of use. This was captured with the Treatment Satisfaction Questionnaire for Medication (TSQM; version 1.4) on a 7-point scale, ranging from extremely difficult to extremely easy and was based on last post-dose observation (n=154). This set was dichotomized into “difficult” for the categories extremely difficult, very difficult, and difficult; and into “easy” for the categories somewhat easy, easy, very easy, and extremely easy. Using this approach, 88% of patients rated the technique of SCIg administration easy to learn.52
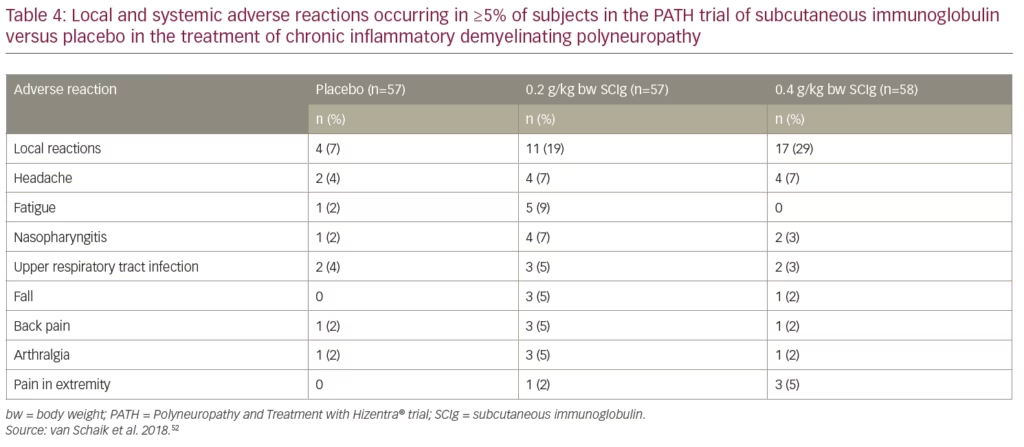
Patients stabilized on IVIg reported a 3.6-fold lower rate of systemic adverse reactions per infusion after they were switched from IVIg to SCIg.52,61 Please see Table 4 for a list of adverse reactions occurring in greater than or equal to 5% of subjects who received Hizentra® or placebo. Local reactions, headache, fatigue, and nasopharyngitis were increased with SCIg compared with placebo and the majority of events were considered mild to moderate. Adverse reaction rates per infusion for 0.2 g/kg bw SCIg, 0.4 g/kg bw SCIg, and placebo, were: 7.9%, 5.1%, and 3.4%, respectively.49,61 Patients reported that the most common local reactions were erythema and swelling; most were considered mild to moderate; frequency and severity decreased over time.
Overall, primary and secondary endpoint findings together with other analyses of the PATH study have provided evidence that SCIg is an effective alternative to IVIg in CIDP maintenance therapy with fewer reported systemic adverse reactions per infusion.
Discussion and future directions
CIDP is a disease that may require long-term maintenance therapy and can be effectively treated and stabilized with immunoglobulin therapy. The evidence from various studies, particularly the pivotal PATH trial, indicates that SCIg is effective as maintenance therapy in adults with CIDP.52 This efficacy is similar to that seen with IVIg in previous studies. Advantages over placebo were shown in the primary and secondary endpoints of the PATH study and in other analyses. Treatment with SCIg resulted in fewer reported systemic side effects per infusion than IVIg, and does not require venous access. Clinical study data are now available that support the use of SCIg therapy for up to 18 months. Maintenance therapy beyond 18 months should be individualized based upon the patient’s response and need for continued therapy.49
An advantage of SCIg is that dosing can be more flexible and more frequent than IVIg. Flow rates, infusion sites, and site volumes can be readily altered as needed. The recommended dose of SCIg is 0.2 g/kg bw (1 mL/kg bw) per week. In the PATH study after transitioning from IVIg to SCIg, a dose of 0.4 g/kg (2 mL/kg) bw per week was also safe and effective to prevent CIDP relapse. If CIDP symptoms worsen, consider re-initiating treatment with an IVIg approved for the treatment of CIDP, while discontinuing SCIg. If improvement and stabilization are then observed, consider reinitiating SCIg at the dose of 0.4 g/kg bw per week, administered in two sessions over 1 or 2 consecutive days, while discontinuing IVIg. If CIDP symptoms worsen on the 0.4 g/kg bw per week dose, consider re-initiating therapy with an IVIg product approved for treatment of CIDP, while discontinuing SCIg.49 The starting dose and subsequent dose adjustments are subject to physician judgement, relapse control, and tolerability. The benefits of self-administration at home and the lower incidence of systemic adverse effects could be beneficial to patients for whom IVIg presents a treatment or lifestyle burden.
Questions and answers on the use of subcutaneous immunoglobulin (Hizentra®) in chronic inflammatory demyelinating polyneuropathy
Question: Which patients with CIDP are suitable for treatment with Hizentra® (SCIg)?
Answer: Adults whose CIDP is stable with IVIg treatment but have issues or difficulties with such treatment can be considered for switching to SCIg. These issues include:
- Inconvenience of infusion clinic or home nurse visits (due to work, lifestyle, or caregiver schedules)
- Lack of reliable access to infusion clinic or home nurse visits (due to location, lack of transport or insurance not covering home visits)
- Poor venous access, higher risk for IV-related adverse reactions (especially in patients with ports)
- Requiring more frequent infusions to manage disease
- Systemic adverse reactions on IVIg (e.g. headache and nausea)
- Preference for independence.51,59
Question: How and when is a patient started on SCIg? What dose should be given; can the dose be changed as a result of efficacy or tolerability issues? Can the SCIg dose be titrated?
Answer: A patient can be started on SCIg if CIDP is stabilized with IVIg and the physician considers a patient to be suitable for SCIg treatment. Dosing and administration should adhere to the Hizentra® Prescribing Information, which describes infusion procedures in detail.49 The recommended dose of SCIg in CIDP is 0.2 g/kg bw/week administered in 1 or 2 sessions over 1 or 2 consecutive days. In the clinical study, after transitioning from IVIg to SCIg treatment, a dose of 0.4 g/kg bw/week was also safe and effective to prevent CIDP relapse. If CIDP symptoms then worsen, re-initiating treatment with an IVIg approved for the treatment of CIDP, while discontinuing SCIg should be considered. If there is improvement and stabilization during IVIg treatment, reinitiating SCIg at 0.4 g/kg bw/week in 1 or 2 sessions over 1 or 2 consecutive days, while discontinuing IVIg can be considered. If CIDP symptoms then worsen on the 0.4 g/kg bw/week dose, re-initiating therapy with IVIg, while discontinuing SCIg should be considered. The patient’s clinical response should be monitored and the duration of therapy adjusted based on patient need.
In CIDP, a first SCIg infusion of ≤20 mL/site at a rate of ≤20 mL/hr/site is recommended. Subsequent infusions can be ≤50 mL/site at a rate of ≤50 mL/hr/site, as tolerated. An example is a 165 lb (75 Kg) adult with CIDP who receives 15 g/75 mL, starting on SCIg at 0.2 g/Kg bw and would infuse at 20 mL/hr/site. Therefore, this patient would need to infuse at four sites which would take approximately 1 hour.49
Question: Is SCIg administration easy for the patient or caregiver?
Answer: In the PATH study, patients’ ease or difficulty in learning the technique of self-administration of SCIg were rated as either ‘difficult’ (categorized as: extremely difficult, very difficult, and difficult) or ‘easy’ (categorized as: somewhat easy, easy, very easy, and extremely easy). Among patients, 88% rated SCIg as one of these four categories of easy to learn.52 This was an exploratory endpoint based on the last post-dose observation (n=154) captured using the TSQM version 1.4. All patients or caregivers learned to effectively administer SCIg on their own after ≤4 training sessions.
Question: How long can a patient receive SCIg treatment?
Answer: Maintenance therapy with SCIg in CIDP has been systematically studied for 6 months and for a further 12 months in a follow-up study. Maintenance therapy beyond these periods should be individualized based upon the patient’s response and need for continued therapy.49
Question: Can a patient receive SCIg who has not received previous CIDP treatment?
Answer: Treatment with SCIg is currently approved in CIDP only as a maintenance therapy for patients whose disease has been controlled using IVIg, so is not suitable for patients who have received no prior treatment. It is recommended that SCIg therapy is initiated 1 week after the last IVIg infusion.
Question: What adverse reactions are associated with SCIg treatment?
Answer: In the PATH study, the most common adverse reactions observed in ≥5% of clinical study subjects receiving SCIg were local reactions (e.g. pruritus, hematoma, erythema, swelling, pain, induration, heat at the infusion site). Systemic reactions (≥5%) include: fatigue, headache, arthralgia, back pain, pain in extremity, nasopharyngitis, and upper respiratory infection.49,52 It should be noted that all immunoglobulin products carry a risk of thrombosis.
A patient’s journey with chronic inflammatory demyelinating polyneuropathy
Patient narrative
A 25-year-old female nursing school attendee experienced weakness in her legs and difficulty in grasping, reaching and standing. After 3 months of these symptoms, on one occasion, she needed assistance rising from a seated position. This prompted a visit to her primary care physician who identified neurological deficits. She was referred to a neurologist who made a presumptive diagnosis of Guillain-Barré syndrome.
Her symptoms were stable for 6 months but had worsened by the time of a 12-month follow-up visit and the diagnosis was changed to CIDP. She was prescribed concomitant therapies including a course of IVIg treatments (five treatments within 2 weeks). She reported symptom improvement as regained feelings in her fingers, but after 5–6 weeks the symptoms returned. The patient was offered corticosteroids as an alternative option but declined, being aware of the side effects associated with long-term treatment. She then consulted a CIDP neuromuscular specialist who prescribed IVIg every 3 weeks.
Although the patient experienced an improvement in symptoms, there were systemic adverse reactions, particularly diarrhea and headaches. There were also problems achieving venous access despite good hydration; placement of a central venous catheter was discussed. Additionally, the IVIg treatments were associated with several logistical burdens including regularly missing work due to travel time to the clinic and the duration of each IV infusion being 6–8 hours.
The neuromuscular specialist suggested enrolling in a clinical trial of Hizentra® as a maintenance therapy. After switching to this treatment, she found at-home administration very convenient giving her flexibility and control over her treatment and avoiding the increasing difficulties with venous access. The patient no longer reported headaches following treatment, although mild and manageable diarrhea persisted. She found self-administration to be a very straightforward process and was confident with the technique after two sessions (It should be noted that in the PATH study, all patients or caregivers learned to effectively administer SCIg on their own after ≤4 training sessions). The patient has a nursing background but non-medical patients can also be trained, and further training and support are available. In those who are unable to self-administer, a caregiver or a nurse can assist. The SCIg infusion duration provided a beneficial interval in her routine when she could relax, watch TV or read. The infusion pump can fit inside loose clothing or can be placed in a bag enabling some movement and with approval from her physician, certain tasks can be carried out while the infusion is in progress.
In her hospital working environment, the patient is able to inform other patients with CIDP about CIDP and available treatments based on her personal experience. Overall, the patient thought that self-administration of SCIg at home was highly advantageous due to the fact that it fit better into her lifestyle.
Physician comment
This testimony emphasizes that CIDP is a chronic disease with variability in presentation and progression and correct diagnosis is frequently delayed.2,62 It also supports study evidence that IVIg helps fulfill patient needs. IVIg treatment, however, is often associated with flu-like symptoms, headache, nausea, diarrhea, and fatigue.33,35,58
The testimony also highlights the main reasons why patients decide to switch to SCIg from IVIg, which include: seeking greater control and freedom, having poor venous access, experiencing IVIg-related side effects or requiring more frequent infusions to manage the disease.33,51 When managing patients with CIDP who are receiving IVIg treatment, it is important to regularly ask them about their condition and the treatment effects. If patients have issues with this treatment, such as headaches, the physician could then recommend alternative options such as a trial switch to SCIg.
With SCIg, this patient reported some diarrhea but had fewer or no headaches or nausea which accords with experiences of other patients who switched to SCIg. The patient also found that self-administration at home was convenient and easy to learn. Although she is a nurse and experienced, patients unfamiliar with the treatment can be trained and are offered continued support during their treatment. The dosing calculator on the Hizentra® website and the app63 help physicians correctly determine what dose to administer to each patient depending on patient criteria.