Narcolepsy is a chronic clinical condition primarily characterized by excessive daytime sleepiness (EDS). This may be accompanied by cataplexy, which is a phenomenon of transient muscle weakness triggered by strong emotions, such as laughter, excitement, anger or grief. Narcolepsy is subtyped as type 1 (NT1) if cataplexy is present or type 2 (NT2) if cataplexy is absent. Other symptoms include disturbed nighttime sleep, sleep paralysis, and hypnagogic and hypnopompic hallucinations, all of which can significantly affect quality of life (QOL). On a global scale, NT1 affects 25–50 people per 100,000, with an incidence of 0.74 per 100,000 person-years.1 While the exact prevalence of NT2 is uncertain, it has been estimated to be 20–34 per 100,000 persons in one community setting.2 A more recent study conducted in the United States stated a prevalence of 44.3 per 100,000 persons as of 2016.3
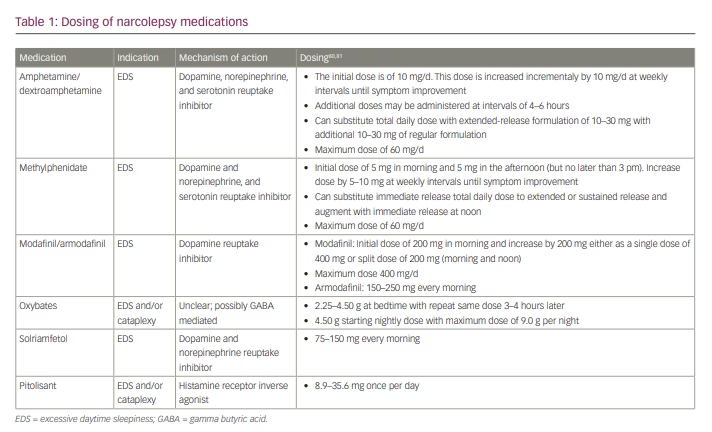
Treatment of narcolepsy focuses primarily on QOL improvement via reduction of EDS, maintenance of nocturnal sleep, and mitigation of other narcolepsy symptoms, including cataplexy, sleep paralysis and hypnagogic/hypnopompic hallucinations. Non-pharmacologic therapy emphasizes regular and sufficient nocturnal sleep and strategic timing of daytime naps to enhance wakefulness.4,5 When selecting pharmacologic therapeutic options, it is important to consider whether the major clinical symptom is EDS, fragmented nocturnal sleep, cataplexy or all of these. Factors such as comorbidities, development of tolerance to long-term medication, and the potential for medication abuse must also be considered. Furthermore, as combination therapy is often required, it is important to be mindful of medication interactions. In this paper, we review the available pharmacologic treatments for narcolepsy, and discuss agents currently under investigation. Specific dosing of agents discussed in this review are listed in Table 1.
Amphetamines and methylphenidate
The most frequently reported symptom of narcolepsy is EDS.6,7 Stimulants such as amphetamines and methylphenidate were among the first available agents to address EDS before newer agents discussed later in this review.8 These medications increase noradrenergic, dopaminergic and serotonergic (only for amphetamines) activity by inhibiting their reuptake and increasing their synaptic concentrations to promote alertness. Wakefulness may also be promoted by increased amine signalling through direct effects on the cortex or via subcortical pathways. However, these drugs can be addictive and there is the potential for abuse.9 Furthermore, higher doses have been associated with adverse effects leading to hospitalization, including cardiac arrhythmias and psychiatric disturbances.9 Evidence for this medication as sole or primary therapy is lacking.
Modafinil and armodafinil
Due to the potential adverse effects associated with amphetamines and methylphenidate, modafinil and armodafinil are the preferred first-line, wakefulness-promoting agents.10 Modafinil and armodafinil block activity of the dopamine transporter, increasing synaptic dopamine, which leads to augmented stimulation of dopaminergic receptors.9,11 It has been reported that these medications also have activity on noradrenergic and serotonergic pathways, but their inability to mitigate cataplexy likely suggests that this activity is negligible.8 Modafinil has the benefits of stimulants with a lower abuse potential.12 Armodafinil is the R-enantiomer of modafinil, and has a longer duration of action. Though armodafinil’s half-life is the same as that for modafinil, it is metabolized in a monophasic manner, resulting in a slower initial clearance compared with the biphasic manner of modafinil.13
To our knowledge, there have not yet been any trials comparing the efficacy and adverse event profile of modafinil against armodafinil in the treatment of narcolepsy. Clinically, choosing between the two formulations may depend on the patient’s timing and duration of EDS. Armodafinil may be more suitable for patients who have an earlier daily onset and/or longer duration of EDS. Headaches are the most common side effect with modafinil and armodafinil, followed by gastrointestinal side effects, including decreased appetite, nausea and abdominal pain.14,15 Stimulants, in general (including amphetamines), may also increase heart rate and blood pressure, with risk of hypertension.16
Improvement in QOL has been reported with both modafinil and armodafinil. In a 9-week, placebo-controlled, double-blind study, modafinil 400 mg showed improvement in the 36-item Short-Form Health Survey (SF-36), a self-administered questionnaire concerning health-related QOL.17 The treatment group reported more energy, fewer difficulties performing usual activities, less interference with social activities, and increased productivity, attention and self-esteem compared with placebo.17 Similar results were also shown in a 6-week, open-label, multicentre study of modafinil for the treatment of narcolepsy.18 While armodafinil has not been as robustly studied in the context of narcolepsy, patients’ improved ability to engage in daily activities has been shown.19
A recent trial, conducted in France, tested the combination of modafinil and flecainide compared with modafinil alone in sleep-deprived subjects.20 This was based on the interaction of these medications with neuroglial connections via connexons, and demonstration of improvement in wakefulness and cognition with this combination in animal models.21 The study showed that psychomotor vigilance, working memory and executive functions were improved compared with modafinil alone. This was followed by a clinical trial that examined the safety and efficacy of this combination versus modafinil alone. However, preliminary results did not show any significant difference in EDS measured by change in Epworth Sleepiness Score (ESS).22
Oxybates
Sodium oxybate is a metabolite of gamma aminobutyric acid (GABA) that acts as a neurotransmitter and neuromodulator. Its mechanism of action is thought to be mediated by GABAB receptor activity at noradrenergic, dopaminergic and thalamocortical neurons, as well as at specific gamma-hydroxybutyrate (GHB) receptors.5 It is widely used to treat narcolepsy due to its effectiveness in suppressing cataplexy, with the additional benefits of improvement of EDS and consolidation of sleep. It is worth noting that improvement in EDS with sodium oxybates may take up to 8 weeks.4 Sodium oxybate is a liquid formulation taken in two doses each night. The starting dose is 2.25 g at bedtime, followed by another 2.25 g 4 hours later, totalling 4.50 g each night. The dose may be increased by 1.5 g per night at weekly intervals based on efficacy and tolerability, up to a maximum dose of 9 g per night.23
Sodium oxybate can be used as monotherapy but is often combined with stimulants. Subjective and objective measures of sleepiness have been shown to improve with a combination of modafinil and sodium oxybate compared with modafinil alone.24 QOL improvement has also been shown by improvements in Functional Outcomes of Sleep Questionnaire (FOSQ) scores across 4 weeks of stable dosing, and SF-36 across 8 weeks after initiation.25–27
Common side effects of sodium oxybate include nausea, mood swings and enuresis, which show increasing incidence with dose escalation and subsiding with discontinuation.28 In addition, non-rapid eye movement sleep parasomnias, such as somnambulism, can occur with sodium oxybate. Sodium oxybate is a central nervous system (CNS) depressant, and while data regarding the impact on obstructive sleep apnoea (OSA) is mixed, some reports indicate worsening of OSA with treatment, and there are reports of central sleep apnoea with sodium oxybate. Therefore, caution must be exercised when sodium oxybate is prescribed in the setting of comorbid sleep-disordered breathing.10,29,30
Sodium oxybate has a high salt content, which may be a limitation for use in patients who are sensitive to salt intake such as those with hypertension, heart failure, or fluid overload in the setting of renal failure. The maximum dose of 9 g per day contains 1,647 mg of sodium. This is approximately 700 mg below the upper limit of sodium intake recommended by the American Heart Association for patients with hypertension.31 A newer oxybate formulation replacing the majority of sodium with magnesium, calcium and potassium, reduces the sodium content by approximately 90%.32 Despite the higher magnesium and potassium content, dose adjustments are not required for patients with renal failure and, to our knowledge, there are no reports of increased complications from hyperkalaemia or hypermagnesemia.33
Contraindications include concomitant use with alcohol or other sedative-hypnotic medications. Sodium oxybate is also contraindicated in a rare autosomal recessive disease, succinic semialdehyde dehydrogenase deficiency. Succinic semialdehyde dehydrogenase is necessary for the breakdown of GABA and GHB, and its deficiency leads to increased concentrations GABA and GHB, which can result in seizures.34 While narcolepsy is not reported to be common in this rare disease, one study showed decreased mean sleep latency in 5 out of 10 patients (on 6 out of 11 multiple sleep latency tests [MSLTs]), suggestive of hypersomnolence with 2 sleep-onset rapid eye movement episodes seen in 2 paediatric patients.35 Drug abuse/misuse is also of concern; however, these medications are distributed through a central pharmacy, which has kept these incidents rare.36 Withdrawal symptoms from sodium oxybate therapy in the context of narcolepsy have not been formally reported.37 However, withdrawal symptoms including delirium, depression and anxiety, from illicit use of GHB at daily doses less than
10 g have been reported.38,39
The currently available formulations of oxybate require an inconvenient dosing schedule, in which the two doses are split 4 hours apart as discussed above.23 Patients will either awaken spontaneously or set an alarm to take the second dose. In the authors clinical experience, some patients have achieved adequate clinical improvement with only the bedtime dose of sodium oxybate. To eliminate the need to awaken for the second dose, a controlled-release formulation of sodium oxybate (FT218) is currently being studied in a phase III trial (NCT04451668).40 This formulation uses a microparticulate platform that can deliver orally administered small-molecule medications in both delayed and/or an extended manner. Enrolment for this trial included patients with either NT1 or NT2, and primary outcome measures are maintenance of wakefulness test (MWT) sleep latency, Clinical Global Impression of Change (CGI-C) sleepiness scores, and mean number of cataplexy attacks. Compared with placebo, patients who received FT218 showed improvements in all of these outcomes.40
Solriamfetol
Solriamfetol is a dopamine and norepinephrine reuptake inhibitor used to improve wakefulness in narcolepsy and OSA. It increases the extracellular concentration of dopamine and norepinephrine in the striatum and prefrontal cortex, resulting in alerting effects. In a recent international, double-blind, randomized, placebo-controlled, phase III trial, solriamfetol showed a dose-dependent improvement in MWT and ESS score, with 150 mg and 300 mg doses meeting the MWT and ESS coprimary endpoint.41 A recent systematic review compared solriamfetol, modafinil and armodafinil against placebo to examine the improvement in ESS, MWT and patient CGI-C in patients with OSA and EDS. Although not a direct comparison, the study concluded that solriamfetol had the highest probability of improvement in all three outcomes.42 Favourable results have been suggested, including a study by Weaver et al. which analysed the long-term effects of solriamfetol on QOL in participants with EDS associated with narcolepsy or OSA. The FOSQ-10, an abridged version of FOSQ, and SF-36 overall improved in their cohort of
226 patients with narcolepsy. The magnitude of improvement was found to be similar for the 417 patients with OSA that were also included in this study.42–45 It must be noted that solriamfetol does not have any effect on cataplexy, and should not be used as monotherapy in patients with NT1.
Adverse effects have been recorded with an incidence of ≥2% compared with placebo, with the most common reported effects being headache, decreased appetite, nausea, anxiety, insomnia, dry mouth, constipation and palpitations.10 Dose-dependent increases in blood pressure and heart rate have also been reported. However, in the phase III study, these increases seem to be minimal, with the mean change from baseline to week 8 in systolic blood pressure being only 1.6–2.4 mmHg across doses of 75–300 mg/day. Diastolic blood pressure and heart rate increased by 1.0–3.0 mmHg and 0.2–4.8 beats per minute, respectively.46 However, concurrent use of monoamine oxidase inhibitors (MAOI) may increase the risk of hypertensive reaction; therefore, MAOI use is contraindicated with solriamfetol or within the preceding 14 days of solriamfetol initiation.47 Furthermore, a long-term study of the safety and maintenance of efficacy of solriamfetol in participants with narcolepsy or OSA showed a higher prevalence of cardiovascular serious treatment-emergent adverse events (TEAEs) in patients with OSA; however, this is likely due to the high prevalence cardiovascular comorbidities in these patients.44
Pitolisant
Pitolisant is a histamine 3 receptor antagonist/inverse agonist that improves both EDS and cataplexy in narcolepsy. Its effect is thought to be mediated pre-synaptically through activity on histaminergic neurons in the brain. It enhances histamine release in the CNS by blocking the inhibitory effect of histamine on its own endogenous release.8 Animal models have also shown that it increases the release of acetylcholine and dopamine in the cerebral cortex, but not in the striatal complex, which is thought to partly explain its lower abuse potential.48
Clinical trials of pitolisant have shown favourable responses in reducing sleepiness. A recent systematic review, with a primary outcome of change in ESS score, suggested a reduction is ESS score of 5.9–6.2 points from baseline.49 A separate analysis of data from two placebo-controlled studies, one of 7 weeks’ duration and one 8 weeks, both permitting titration, showed that mean reductions in EDS were significantly greater relative to placebo, even after 2–3 weeks of treatment.50 A double-blind randomized trial (Harmony 1) compared pitolisant with placebo for superiority, and pitolisant with modafinil for non-inferiority, based on ESS score change from baseline.40 The results showed superiority of pitolisant versus placebo; however, while there was no significant difference between pitolisant and modafinil, the mean difference was outside the pre-specified non-inferiority margin. Therefore, non-inferiority of pitolisant to modafinil was not shown. Fewer adverse events were recorded in the pitolisant group compared with modafinil.51 Though studies examining its QOL outcomes are limited, improvements in CGI-C and European Quality of life questionnaire (EQ-5D) have been reported.52,53
Pitolisant has been shown to reduce cataplexy in clinical trials. In a 7-week, randomized, double-blind trial, pitolisant decreased weekly cataplexy rate by 75% in those who experienced at least three cataplexy attacks per week. This decrease from baseline was significantly greater with pitolisant (from 9.1 to 2.3 attacks per week; -75%) than with placebo (from 7.3 to 4.5 attacks per week; -38%), and was seen between the 2-week baseline period and the 4-week stable-dose period (p=0.004).54 A post hoc analysis included in an earlier trial showed that 61% of patients had a reduction in daily rate of cataplectic attacks with pitolisant compared with placebo (0.18 versus 0.39 attacks per day).51 A recent study investigating the long-term safety and efficacy of pitolisant, was a 12-month, pragmatic, open-label, multicentre study (Harmony 3).52 Mean total cataplexy episodes per day decreased by 68% compared with baseline. Reductions in hypnagogic hallucinations and sleep paralysis were also reported. However, 35% of participants were taking other narcolepsy medications (stimulants, sodium oxybate, antidepressants). Furthermore, only 68 of the 102 participants who received pitolisant completed ≥12 months of treatment. Discontinuations mostly occurred during the first 3 months, with perceived insufficient efficacy and adverse events being the most common reason for discontinuation.52
Pitolisant is generally well tolerated but does have significant adverse effects. Scart-Grès et al. recently analysed data from four randomized, placebo-controlled trials evaluating adverse events, vital signs, laboratory tests and electrocardiogram (ECG) measurements in 303 adult patients with narcolepsy (pitolisant n=172; placebo n=131).55 Overall, the incidence of adverse events was 49.4% with pitolisant compared with 41.2% with placebo. The most common adverse events for pitolisant versus placebo were headache (18.0% versus 13.7%), nausea (5.2% versus 3.1%), insomnia (4.1% versus 2.3%), upper respiratory tract infections (4.1% versus 0.8%), back pain (3.5% versus 0.8%) and dizziness (3.5% versus 2.3%). Adverse events resulted in treatment discontinuation for 3.5% and 3.8% of patients in the pitolisant and placebo groups, respectively. Changes in vital signs, laboratory tests and ECG parameters attributable to pitolisant were not seen.55
In the abovementioned Harmony 3 study, 57% of participants treated with pitolisant reported TEAEs, the majority of which (55%) occurred during the first 3 months.52 A higher rate of TEAEs was seen in patients who were receiving concomitant narcolepsy treatment. Reported TEAEs were similar to the adverse events reported by the Scart-Grès et al. study et al, and included headaches (11.8%), insomnia (8.8%) and nausea (4.9%).55 Harmony 3 also reported weight gain (7.8%), anxiety (6.9%) and depression (4.9%).52
Drug–drug interaction studies have shown that CYP3A4 inducers reduce pitolisant exposure, and CYP2D6 inhibitors increase pitolisant levels.56 Dose reductions are thus recommended with concomitant use of CYP2D6 inhibitors and dose increases with strong CYP3A4 inducers. Caution is advised when using pitolisant in combination with drugs that interact with these enzymes, including immunosuppressants, docetaxel and kinase inhibitors. Pitolisant may also decrease the efficacy of oral contraceptives, and females of reproductive potential should be advised to use non-hormonal contraceptive treatments when taking pitolisant and for ≥21 days after the last dose.8 It is also contraindicated in those with severe hepatic impairment (Child–Pugh class C).56 Furthermore, pitolisant can interact with QT prolonging agents, such as haloperidol, and increase the risk of ventricular arrhythmias.44
Some of pitolisant’s side effects are attributed to its nonspecific binding to the sigma-1 and sigma-2 receptors. While not fully understood, these receptors are known to produce anxiogenic effects and are thought to be a risk factor for depression and addiction.57,58 Furthermore, drug–drug interactions can limit pitolisant use due to its interaction with the cytochrome P450 (CYP) system.56 Unlike pitolisant, samelisant (SUVN-G3031), a new H3 inverse agonist currently being tested in a phase II clinical trial (NCT04072380), has no significant binding affinity at sigma-1 and -2 receptors, and reportedly, has no inhibition or induction of major CYP enzymes.58,59 In animal models, samelisant caused significant increases in acetylcholine, histamine, dopamine and norepinephrine levels in the cortex but not in the striatum and nucleus accumbens, which may indicate a lower abuse potential.58 Preclinical studies of samelisant have indicated no negative effects on ECG, fertility or foetal development, and no CNS safety issues.60 While its efficacy, especially compared with pitolisant, is unknown, it could be a useful agent that may expand therapeutic options for narcolepsy.
Rapid eye movement suppressants
Prior to the development of the above therapy options, antidepressants were commonly used for treatment of cataplexy. Tricyclic antidepressants, including clomipramine, imipramine and desipramine, were the first medications used for this purpose. Subsequently, selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors have replaced tricyclic antidepressants, with venlafaxine frequently prescribed for the treatment of cataplexy.11 However, there are insufficient data to support all rapid eye movement (REM) suppressant medications as a first-line therapy, and with newer available therapeutics, these are now second-line agents. There are also concerns of side effects from REM suppressants, including constipation, dizziness, dry mouth, urinary disturbance, anxiety and insomnia.61 REM suppressants may also precipitate other sleep disorders such as REM behaviour disorder and restless leg syndrome.62,63 With abrupt withdrawal of any of these medications, cataplexy rebound can also happen.64
Another norepinephrine reuptake inhibitor, reboxetine, recently completed a phase II trial assessing the long-term efficacy for treatment of cataplexy and EDS associated with narcolepsy (NCT05113745). Reboxetine was originally developed for treatment of depression.8 Preclinical animal models have shown reduction in cataplectic episodes, and a 2-week pilot study in 12 patients showed a reduction in ESS by 49% and an increase in MSLT sleep latency by 55%.65,66 The pilot study reported adverse events, including dry mouth, hyperhidrosis, constipation and restlessness. Post-marketing data for those treated with reboxetine for depression reported insomnia, dizziness and nausea.67
Orexin receptor antagonists
The pathophysiology of narcolepsy includes the destruction of hypocretin/orexin neurons, leading to decreased orexin levels. Consequently, orexin-based pharmacologic treatments have been of interest, and are currently under investigation. Two forms of orexin receptor agonists have been studied so far: TAK-925 and TAK-994. TAK-925 is administered subcutaneously, and has demonstrated improved wakefulness, reduced cataplexy episodes and ameliorated weight gain in animal models.68 A separate animal model has also shown that TAK-925 enhanced cortical arousal and suppressed signs of somnolence and drowsiness in comparison with modafinil.68,69 Phase I data for TAK-925 in individuals with NT1 showed increased maintenance of wakefulness and MSLT scores, and lower Karolinska Sleepiness Scale scores. In this study, incidence of TEAEs was chosen as the primary endpoint. In total, 21.7% of patients in the placebo group reported at least one TEAE, compared with 32% in the lower-dose and 41.7% in the higher-dose TAK-925 groups. No serious TEAEs or discontinuation due to TEAEs were reported, and all TEAEs were recorded as mild to moderate in severity (NCT03748979).70,71.53,54
TAK-994 was developed more recently as an oral medication. It has also shown similar results to TAK-925 in animal models, as well as ameliorating the fragmentation of wakefulness.72,73 Unfortunately, a phase II trial of TAK-994 has recently been halted due to safety concerns. While the exact reason for discontinuation is unclear, there is some suggestion that this was due to hepatotoxicity.74,57
Another medication currently being studied as an orexin agonist, is mazindol. Mazindol is a tricyclic anorectic, non-amphetamine stimulant that has been used for narcolepsy since 1970. While there were reported benefits of mazindol for both EDS and cataplexy, there have not been any clinical trials investigating its use in patients with narcolepsy.75 In November of 2010, a different anorectic drug was reported to cause valvulopathies and pulmonary hypertension, leading to a re-evaluation of the safety profile of mazindol.75 Due to the lack of therapeutic data on the potential side effects, it has fallen out of favour and has been withdrawn from the market.
In addition to its activity on monoamine reuptake, mazindol’s effect on narcolepsy symptoms is mediated by its orexin agonist effect, particularly through the OX2R receptor. A recent pharmaceutical company-sponsored preclinical study, conducted at the University of Lausanne in Switzerland, reported that OX2R knock-out mice showed 70% decreased sensitivity to mazindol compared with wild-type mice.76 A new controlled-release formulation is in development, which may decrease the rate of adverse events formerly associated with mazindol. A phase II, 4-week, double-blind, placebo-controlled, randomized, multicentre, parallel-group study to assess its efficacy and safety is currently ongoing (NCT04923594). Orexin is undoubtedly an important component of narcolepsy pathophysiology, and while further studies are needed, it is an important pharmaceutic target.
Immunotherapy
Depletion of orexinergic neurons is thought to be responsible for many of the symptoms observed in patients with NT1. While the pathophysiology of NT2 is less clear, it has been suggested that there is a less-severe loss of orexinergic neurons or other alterations in the orexin system compared with NT1.77 Studies have shown a strong association between the loss of orexinergic neurons and HLA-DQB1 *0602 gene expression.77 Infections can also be related to NT1 incidence through molecular mimicry. This was particularly apparent following a vaccine campaign (Pandemrix; GlaxoSmithKline, Brentford, West London, UK) during the 2009 H1N1 pandemic. It is believed that the haemagglutinin protein triggers a T-cell- mediated autoimmune process resulting in decreased orexin activity.This led to the hypothesis that there may be an autoimmune component within the hypothalamus, leading to the development of narcolepsy.77,78 Immunotherapy has been studied in patients with narcolepsy in the hope of developing symptomatic treatment from this autoimmune standpoint. Small case series and case reports exist on the use of intravenous immunoglobulins, plasma exchange, systemic corticosteroids and monoclonal antibodies. However, none of these have been sufficient to support its use, and are limited by their uncontrolled design and small sample size.79 Currently there is no evidence for use of these interventions.
Conclusion
Narcolepsy is a debilitating chronic condition that can be challenging to treat. With expanding knowledge of the pathophysiology of narcolepsy and the neurobiology of the CNS wake and sleep regulatory systems, significant advancements have been made in pharmacologic therapy for this disorder. While there is a paucity of head-to-head trials and limited comparison due to differences in utilized scales, these medications can improve patients’ QOL. However, therapy is still focused on symptom management, and a cure remains elusive. The advent of personalized medicine and advances in pharmacogenomics may lead to further approaches targeting the autoimmune process that is believed to be the basis of this disorder, as well as restoring hypothalamic orexin function.