The same basic regulations from the US Code of Federal Regulations (CFR) apply to all clinical investigations of medical devices. These include 21 CFR Part 50 (informed consent), Part 54 (financial disclosure), Part 56 (institutional review boards (IRB)), and Part 812 (investigational device exemptions (IDE)). The IDE regulation:
• stipulates that all studies must be approved by the FDA and IRB before they begin;
• assigns responsibilities to sponsors and investigators; and
• mandates written informed consent for all study subjects.
Device studies conducted to support a US marketing application, or those involving collection of safety and effectiveness information for a new intended (‘offlabel’) use of a marketed device (including studies conducted by ‘sponsor-investigators’ rather than a manufacturer), are all subject to these regulations. Certain clinical studies are exempt from this regulation, including those of devices commercialized prior to 1976 (when the Medical Device Amendments Act was implemented), devices used in studies in accordance with their approved labeling, most studies of in vitro diagnostic devices, consumer preference testing of marketed devices, combinations of legally marketed devices, custom devices, and studies conducted outside the US. Such studies are generally still subject to the informed consent requirement and IRB oversight. If a study is subject to the regulation, then the IRB must determine whether the study is a ‘significant’ or ‘non-significant’ risk. ‘Significant risk’ is defined as “an investigational device that is intended as an implant and presents a potential for serious risk to the health, safety, or welfare of a subject, or is purported or represented to be for a use in supporting or sustaining human life and presents a potential for serious risk to the health, safety, or welfare of a subject, or is for a use of substantial importance in diagnosing, curing, mitigating, or treating disease, or otherwise preventing impairment of human health and presents a potential for serious risk to the health, safety, or welfare of a subject, or otherwise presents a potential for serious risk to the health, safety, or welfare of a subject.”
If a device is determined significant risk, then the sponsor of the proposed study must submit an IDE application to CDRH. This application is approved, conditionally approved (allowing the study to begin), or disapproved within 30 calendar days. Non-significant risk studies are reviewed and approved by the IRB only, but are still subject to ‘abbreviated’ IDE requirements, including informed consent, monitoring, reporting, labeling, and a prohibition on promotion.
Financial disclosure regulations (21 CFR Part 54) require that the financial involvement of investigators be gathered at the time a ‘covered’ study (one that may ultimately support a marketing application) is conducted and updated annually, but submission of this information to the FDA is only required at the time of a marketing application. Financial compensation is not prohibited, nor is divestiture required.
An IDE application can be sponsored by any US entity, including a manufacturer, contract research organization (CRO), academic investigator, or government agency. The IDE sponsor assumes legal responsibility for compliance with all applicable regulations.
CDRH has developed a highly effective ‘pre-IDE’ program to allow for early informal feedback on preclinical testing and clinical protocols before IDE
applications are submitted. It is also useful for nonsignificant risk or exempt studies, and studies conducted outside the US, intended to eventually support a marketing application but that will not have IDEs associated with them. This program provides a collaborative forum for CDRH and sponsors, enabling earlier consensus on device testing and development, increasing the likelihood of approval of an IDE application, and minimizing surprises. Feedback can be in the form of face-to-face meetings, teleconferences, or electronic mail. Pre-IDE feedback is not legally binding on either the FDA or the sponsor. However, there are also binding ‘determination’ and ‘agreement’ meetings designed to reach formal written consensus on the type of data required for a study, as well as study design parameters, for a particular device (see http://www.fda.gov/cdrh/ode/guidance/310.html).
CDRH has a wealth of information available on regulation of clinical studies of medical devices (see http://www.fda.gov/cdrh).
Pre-clinical Engineering Issues
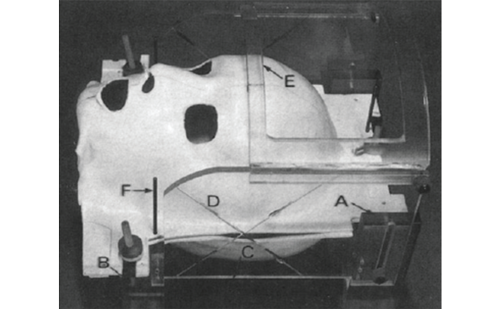
Pre-clinical engineering analyses play a critical role in the development and evaluation of new medical devices, and are much more significant for medical devices than for other types of FDA-regulated products. The medical device industry generates thousands of new products annually. In 2004, CDRH received and processed over 8,000 submissions representing more than 3,000 products, from a diverse range of over 12,000 sponsors and manufacturers, with a similarly broad ranges of size and resources. Many of these medical devices are in a state of frequent design change and evolution compared with FDA-regulated pharmaceutical products. For these and other reasons, clinical trials are neither feasible nor appropriate for many devices. Indeed, thousands of regulatory decisions are made regarding such products each year without clinical trials, relying instead on pre- or nonclinical engineering characterization of device performance. Even for products for which clinical data are required, engineering evaluations often shape the nature of the clinical trials.
While engineering issues are often central in medical device development and approval processes, they pose special technical challenges for manufacturers and for CDRH. Compared with other FDA-regulated areas, medical devices span a dramatic range, from tongue depressors to artificial hearts. Future product types will only intensify these issues. For example, CDRH technology forecasts (see http://www.fda.gov/cdrh/ ost/trends/toc.html) project major pioneering device trends as varied as computerized products, genetic diagnostics, home- and self-care devices, minimally invasive technologies, combination device drug biologic products, and artificial organs.The range of technologies and scientific disciplines involved in the engineering, design, and evaluation of these devices, individually and collectively, is enormous. The expertise required for competent engineering design, testing, and evaluation of medical devices includes not only engineering (e.g., electrical, mechanical, software) but also chemistry, materials science, fluid dynamics, electromagnetics, optics, acoustics, mathematics, toxicology, genetics, and more.
Against this backdrop of widely ranging topics and issues, engineering considerations take several forms for both industry and the FDA. Non-clinical testing often necessitates an initial elucidation of basic mechanisms of action of new technologies and their modes of interaction with the body. Such phenomena underpin the basic safety and effectiveness of devices under actual-use conditions, and point to the types of testing and characterization that are needed. Non-clinical testing for new devices also frequently requires development and validation of new types of test methodologies relevant to new types of devices, in order for engineering data to be reliably interpretable. This test development, validation, and implementation may require application of expertise from many of the areas above. Moreover, sophisticated computational modeling is playing an increasingly important role in device design and performance assessment, and engineering evaluation of a device design frequently comprises a combination of complementary bench and computational testing.
Despite the challenges implicit in these engineering design and evaluation processes, such techniques can ultimately significantly speed innovation by optimizing development and regulatory decision-making for medical devices, and are often vastly less costly than clinical trials.While new device sponsors and designers must unavoidably have considerable expertise of their own, CDRH scientists also facilitate these processes by elucidating and disseminating underlying mechanisms of action in peer-reviewed scientific literature, developing and validating reliable, standardized test methods for new product areas, and contributing these methodologies to national and international consensus standards organizations.These initiatives are targeted to provide a solid scientific foundation for dialogue among stakeholders in new and existing product areas, and are one of the ways CDRH seeks to enable a level playing field for participants in that dialogue.
Human Factors
The field of ‘human factors’ (HF) has become
increasingly important in the development and evaluation of medical devices. Known as ‘ergonomics’ in Europe,HF is a science devoted to the interaction of people and equipment—from simple devices to complex systems and processes.This discipline is applied to the design of the ‘user interface,’ comprised of controls, displays, connections, software, labels, instructions, training, and maintenance—anything the user needs to correctly perform installation, set-up, operation, or repair. The goal is to accommodate the abilities, limitations, and work environments of users, or, in other words, improve ‘user-friendliness.’
While HF has been extensively utilized in the power generation, chemical, and aviation industries for years, medicine has been traditionally slow to apply HF.The objective of HF evaluation of medical devices is to improve human performance and reduce the likelihood of user error and resulting patient injury or death. A 1999 US National Academy of Sciences report stated that medical errors are estimated to cause many deaths in US hospitals annually. It is believed that poor medical device interface design is responsible for many adverse events each year.
The FDA’s Quality System Regulation (QSR; 21 CFR Part 820) requires manufacturers of medical devices to consider user needs, including HF considerations, during different stages of device design and development, in a process known as HF engineering. The definitive stage in this process is design validation. In this phase, manufacturers are urged to consider testing by prospective users with actual production units in realistic scenarios, with the goal of demonstrating safe operation with minimal errors.
These studies should be performed in simulated environments prior to, rather than during, clinical trials because:
• the device needs to be demonstrated as safe in use prior to use with patients;
• clinical trials are frequently performed in an ideal environment with well-trained staff, which is not representative of the typical medical environment once a device is approved for marketing;
• it is unethical to impose hazardous scenarios in the clinical environment;
• performing usability measurements or recordings in a clinical environment may intrude on privacy and patient care; and
• at the clinical trial stage, it may be too late to address identified HF problems with device design changes, and instead often ineffective labeling changes are made to address the problem.
Manufacturers must document these activities in their records (design control files), which are inspected periodically by the FDA.
CDRH’s Human Factors Science and Engineering Branch (HFSEB) strives to reduce the likelihood of dangerous errors related to medical device use through both pre-and post-market activities.The premarket activities are intended to encourage manufacturers to provide high-usability designs in new medical devices. The post-market activities are intended to help the FDA and manufacturers protect the public when medical device use has resulted in adverse events, including injuries and deaths.
CDRH HF experts provide HF information to manufacturers through professional meetings, exhibitions, and issuance of guidance documents, and provide pre-market HF guidance and training to FDA reviewers. When a new device generates concern with dangerous use error, HF experts work with the Office of Device Evaluation (ODE) to review the device design, or the manufacturer’s design and development activities. The manufacturer may refine their device design or consider an improvement in a later version of the device. Examples of such changes include simplified programming, clearer displays, or fail-safe connections.
CDRH HF experts also participate in development of both general medical device standards and specific HF medical device standards. Standards represent consensus among researchers, academia, manufacturers, and regulators on how usability is best achieved.
HF staff also participate in investigations of medical device adverse events with other members of CDRH. HF experts evaluate device design, types of users, reported use environment, and the manufacturer’s proposed solutions.These activities are intended to help the FDA and manufacturers determine whether known adverse events may be the result of errors, and to propose and evaluate possible solutions. Finally, HF experts encourage users and manufacturers to report serious adverse incidents through the mandatory medical device reporting (MDR) system, to facilitate the earliest possible detection of patterns that point to potential problems with the design of the interface of medical devices. For more information, visit the Human Factors Program website (http://www.fda.gov/cdrh/ humanfactors/index.html).