
Lecanemab (BAN2401) is an amyloid-targeting monoclonal antibody currently in phase III clinical trials for prevention of Alzheimer’s disease (AD) in the AHEAD 3-45 study (ClinicalTrials.gov Identifier: NCT04468659)1 and for the treatment of early AD (mild cognitive impairment [MCI] owing to AD, and mild AD dementia) in the Clarity AD study (ClinicalTrials.gov Identifier: NCT03887455).2 In this review, the design and innovative development programme of lecanemab are described, and the changing landscape of AD therapeutics is discussed.
Design of lecanemab
Lecanemab targets epitopes on the protofibrillar form of the oligomeric species of amyloid. Oligomeric amyloid-beta protein (Aβ) is implicated as the most toxic form of this canonical pathology of AD.3 In the range of molecular complexity of Aβ oligomers, the protofibrils are high molecular weight aggregations (>100 kDa) that precede evolution to the fibrils found in insoluble plaque forms of Aβ.4 In vitro, protofibrils adversely impact intracellular processes of neurons.5 A murine monoclonal antibody (mAb158) that targets the protofibrillar species of Aβ was developed by BioArctic. This antibody has 1,000-fold greater selectivity for protofibrils than for Aβ monomers, and is 10–15-fold more selective for protofibrils than for fibrils.6 The humanized version of the murine mAb158 is BAN2401 (lecanemab). The humanized antibody has a binding preference for protofibrils in human brain extracts.3
Genetic evidence supporting protofibrils as a target for anti-amyloid immunotherapy
Rare Swedish families whose members have early-onset familial AD carry the ‘Arctic mutation’. This mutation affects the amyloid precursor protein (APP) and results in accelerated protofibril formation and Aβ fibrillization.7 Fibrillization studies in conditioned media from cells transfected with the Arctic mutation confirm the higher rates of protofibrils formation than other Aβ species.8 Plaques form in patients with the Arctic mutation, and most plaques have a characteristic ring-like structure, unlike the plaques seen in sporadic late-onset AD, suggesting that the protofibril predominance may affect the aggregation properties of Aβ.9 Although both the Arctic mutation and the Swedish mutation have been identified in Swedish families, the former is an APP mutation in the Aβ sequence (E693G), and the latter is a mutation of presenilin 1 in the catalytic subunit of the gamma-secretase complex (PSEN1 H163Y or I43T).9–11
Transgenic mice bearing the Artic and Swedish Alzheimer mutations (tg-ArcSwe) express elevated levels of Aβ protofibrils in the brain, providing an opportunity to study the role of this Aβ species in the biology and behavioural manifestations in this model. Young mutation-bearing mice have high levels of protofibrils and few plaques, whereas older animals have high levels of both protofibrils and fibrillar plaque amyloid. Spatial learning (as assessed by the Morris Water Maze) correlated inversely with protofibril levels.12 The antibody mAb158 reduced brain protofibrils in the tg-ArcSwe mice by up to 42% in an exposure-dependent manner.6
These observations support protofibrils as a target for anti-amyloid monoclonal antibodies. Protofibrils are precursor species to Aβ plaques and occur in sporadic late-onset AD, as well as in the informative mutation-induced forms of AD. Linking a specific genetically informed target to the mechanism of action of a candidate drug increases the likelihood of success in the drug development programme.13,14
Clinical development programme of BAN2401
Based on the evidence of protofibrillar Aβ as a target for AD passive immunotherapy, the success of the mAb158 antibody in targeting the protofibrillar Aβ species, and the availability of the humanized equivalent (BAN2401), phase I single-ascending dose and multiple-ascending dose studies were conducted in the USA between 2010 and 2013 (ClinicalTrials.gov identifier: NCT01230853).15 Participants had mild to moderate AD dementia identified with clinical criteria. The planned outcomes of the trials were the safety, tolerability, immunogenicity, pharmacodynamic response and pharmacokinetics of intravenous infusions of BAN2401. A second phase I study was conducted in Japan (ClinicalTrials.gov identifier: NCT02094729),16 and involved patients with MCI and mild AD dementia.
The US phase I study used a novel staggered parallel single- and multiple-ascending dose design.17 The presence of amyloid-related imaging abnormalities of the effusion type (ARIA-E) or haemorrhagic type (ARIA-H) was assessed with magnetic resonance imaging (MRI). Cerebrospinal fluid (CSF) and plasma samples were analysed to investigate the pharmacokinetics of BAN2401, and effects on biomarkers were studied as pharmacodynamic outcomes. In the single-ascending dose study, 36 patients were randomized in cohorts of six participants to placebo or one of six doses of BAN2401 ranging from 0.1 mg/kg to 15.0 mg/kg. Twenty-four participants in the multiple-ascending dose study were randomized to doses of 0.3–10.0 mg/kg in cohorts of six per dose, and treated for up to 4 months. BAN2401 was infused biweekly. The incidence of ARIA-E/H on MRI was comparable with that of placebo in both single ascending dose and multiple ascending dose cohorts. BAN2401 exposure was dose proportional, with a serum half-life of approximately 7 days. There were no measurable effects of BAN2401 on CSF biomarkers.17 Based on these findings, a phase II efficacy study in early AD was initiated.
The phase II study (ClinicalTrials.gov identifier: NCT01767311)18 was a multinational, multicentre, double-blind, placebo-controlled, parallel-group clinical trial using a Bayesian design with response-adaptive randomization across placebo or five differently dosed active arms of lecanemab to determine clinical efficacy and explore the dose response using a novel composite clinical score, the Alzheimer’s Disease Composite Score (ADCOMS).19,20 The ADCOMS is generated from items of the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-cog), items of the Mini Mental State Examination and all items of the Clinical Dementia Rating (CDR). The ADCOMS was designed to have superior sensitivity to change in patients with early AD.21
A total of 854 participants with early AD and confirmed excessive brain amyloid on positron emission tomography (PET) were randomized to lecanemab or placebo in the Bayesian dose-finding protocol. Doses included 2.5 mg/kg bi-weekly, 5.0 mg/kg monthly, 5.0 mg/kg bi-weekly, 10.0 mg/kg monthly, and 10.0 mg/kg bi-weekly. The early AD population, including patients with MCI due to AD, and mild AD dementia, is now common in clinical trials but was novel at the start of this lecanemab study. Similarly, requiring amyloid positivity to be demonstrated on PET was an innovation that confirmed the presence of the amyloid target in the treatment population.19
The primary endpoint of the phase II trial was Bayesian analysis at 12 months of the clinical change in ADCOMS. Success on the primary endpoint required an 80% probability of ≥25% clinical reduction in decline compared with placebo. Key secondary endpoints included 18-month Bayesian and frequentist analyses of brain amyloid reduction on amyloid PET and clinical decline on ADCOMS, CDR-Sum of Boxes (CDR-SB) and ADAS-cog. Drug–placebo differences in CSF biomarkers and total hippocampal volume using volumetric MRI were assessed.19
At 12 months, the 10.0 mg/kg biweekly dose was selected as optimal in the Bayesian model, and showed a 64% probability of being 25% better than placebo on ADCOMS, missing the 80% threshold for the primary outcome. The study proceeded as planned for a total exposure duration of 18 months. Bayesian analyses for reduction in clinical decline at 18 months compared with placebo for the 10.0 mg/kg biweekly dose showed: a 27% reduction in decline as measured by ADCOMS (with 97.7% probability of being superior to placebo); a 56% reduction on ADAS-cog (with a 98.8% probability of being superior to placebo); and a 33% reduction in decline on the CDR-SB (with a 96.4% probability of being superior to placebo). Applying prespecified frequentist analyses at 18 months demonstrated that lecanemab reduced clinical decline compared with placebo on the ADCOMS by 30% (p=0.034), on ADAS-cog by 47% (p=0.017) and on CDR-SB by 26% (p=0.125) at the 10 mg/kg biweekly dose.19
At 12 months, there were significant and dose-dependent reductions in plaque amyloid in all but the lowest dose group. At 18 months, there were robustly statistically significant reductions in brain amyloid on amyloid PET in all treatment groups, showing dose-dependent decreases with escalating doses of lecanemab. The baseline PET standard uptake value ratio (SUVR) for the 10.0 mg/kg biweekly group was 1.37 with a mean reduction of 0.31 at 18 months, indicating that, on average, participants fell below the threshold for PET positivity by visual read (SUVR 1.11).22 Consistent with PET SUVR results, 81% of participants treated with lecanemab compared with 22% of participants on placebo were amyloid negative by the end of the trial (placebo variability is attributed to inconsistencies associated with amyloid levels near the threshold for a positive visual read in some cases).19
CSF collected from a voluntary subgroup of participants showed significant increases in Aβ42 and significant decreases in phosphorylated-tau (p-tau), but no significant change in total tau compared to placebo. Neurofilament light and neurogranin, markers of neurodegeneration and synaptic degeneration, respectively, trended lower in the active treatment group without reaching statistical significance on most measures. There was a marginally greater decline in hippocampal volume and whole-brain volume with increasing ventricular volume in the active treatment group.19
Lecanemab was well tolerated, with a 9.9% incidence of ARIA-E at 10.0 mg/kg biweekly in the overall population and 14.3% in the apolipoprotein Eg4(APOE-4) carriers compared to 0.8% and 1.1% among those on placebo respectively. A decision by regulatory bodies to not allow randomization of APOE-4 carriers to the 10.0 mg/kg biweekly dose because of concern for ARIA in this participant group occurred soon after trial initiation, resulting in an imbalance in APOE-4 carriers across groups. Combining the two highest doses (the 10.0 mg/kg dose monthly was not affected by the regulatory decision) resulted in attenuated effect sizes that favoured lecanemab.19
Based on the results of the phase II study, lecanemab has entered phase III trials. The Clarity AD study (ClinicalTrials.gov identifier: NCT03887455)2 is similar to the phase II trial and involves amyloid-positive participants with early symptomatic AD. The AHEAD 3-45 trial (ClinicalTrials.gov identifier: NCT04468659)1 of lecanemab involves cognitively normal participants with elevated brain amyloid (A45) or intermediate levels of brain amyloid (A3). All participants must be APOE-4 carriers or have a first-degree relative with onset of dementia before age 75 (or have known elevated amyloid levels from previous assessments). Clarity AD is expected to have its primary completion date in September 2022; AHEAD 3-45 has its primary completion date in October 2027.
Lecanemab has been granted Breakthrough Therapy Designation status by the US Food and Drug Administration (FDA).23 Breakthrough Therapy Designations are intended to expedite the development and review of drugs for serious or life-threatening conditions.24 Breakthrough Therapy Designation requires preliminary clinical evidence that demonstrates that the drug may substantially improve at least one clinically significant endpoint over available therapy. A Breakthrough Therapy Designation ensures more intensive FDA guidance on an efficient drug-development programme, including advising on best approaches for gathering the non-clinical and clinical data necessary for approval; taking steps to ensure that the design of clinical trials is as efficient as practicable; assigning a project lead for the FDA review team to facilitate a timely assessment of the development programme and to serve as a scientific liaison among the cross-disciplinary members of the regulatory team (i.e. clinical, pharmacology-toxicology, chemistry, manufacturing and control, compliance); making an organizational commitment involving senior managers; and providing eligibility for rolling review and priority review. The Breakthrough Therapy Designation will facilitate the regulatory interactions of the lecanemab programme.24
Discussion
Lecanemab has a unique development history (Figure 1). The focus on protofibrils distinguishes this monoclonal antibody from others that are being developed. The phase II study conducted with lecanemab is the largest phase II trial (n=854) conducted in AD.25 Use of the ‘early AD’ concept was a trial innovation when the programme was initiated. Confirming diagnosis of AD biology through amyloid studies at baseline is increasingly recognized as critical to AD disease-modifying trials. Anti-amyloid therapeutics require this to confirm the presence of the target pathology, and non-amyloid-targeting drugs require this to confirm the diagnosis and interpret outcomes. The use of the ADCOMS was a pioneering effort to develop and assess a new composite clinical measure as an outcome for AD trials.21 Bayesian analyses have been used previously in AD trials on a limited basis;26 the lecanemab phase II study used the Bayesian approach effectively in dose selection.19, 20
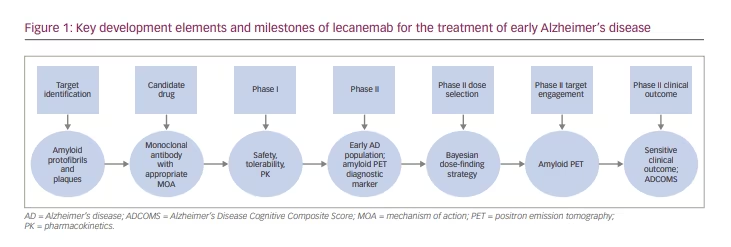
Amyloid PET serves as a target engagement biomarker, as well as a diagnostic confirmation biomarker. Confirming the presence of brain amyloid corroborates the correct diagnosis and ensures that the antibody target is present. The marked amyloid reduction associated with lecanemab treatment provides proof of concept for the protofibrillar/fibrillar targeting and removal by lecanemab. Demonstrating target engagement and proving the intended pharmacodynamics is critical to the success of drug development programmes.27 MRI provides a safety biomarker for monitoring ARIA in lecanemab trials.28
MRI studies showed a decline in hippocampal volume and an increase in ventricular volume in the active treatment group of the phase II trial.19 This has been observed in trials with other amyloid-lowering therapies and is attributed to loss of volume associated with amyloid removal.29,30
Many trials of amyloid-targeting agents have failed to show a drug–placebo difference (as reviewed by Tolar et al., 2020),31 including trials of beta-site amyloid precursor protein cleaving enzyme (BACE) inhibitors whose pharmacodynamics included marked lowering of CSF amyloid monomer levels.30 None of the agents tested in trials with negative outcomes lowered fibrillar/protofibrillar amyloid as visualized on amyloid PET. Amyloid-targeting monoclonal antibodies, including lecanemab, lower fibrillar amyloid levels and have a preliminary signal of clinical efficacy.19,32,34 Reduction in plaque amyloid has been observed in all trials of amyloid-targeting drugs where there is a concomitant suggestion of clinical benefit (e.g. aducanumab, donanemab, lecanemab, gantenerumab).31
The recent accelerated approval of aducanumab by the FDA, based on reduced plaque amyloid revealed by amyloid PET, is ‘reasonably likely’ to predict clinical benefit, and suggests that other amyloid-lowering therapies, such as lecanemab, may be eligible for accelerated approval.35 The approval based on amyloid PET changes and preliminary evidence of cognitive benefit emphasizes the importance of biomarkers for confirming biological effects on the pathophysiology of AD and supporting the treatment as a disease-modifying therapy.
Many types of amyloid imaging, including Pittsburgh Compound B (PIB), are negative in Arctic mutation carriers,36,37 whereas the florbetapir ligand provides a positive image, suggesting that it binds to protofibrils, as well as to the fibrillar amyloid of neuritic plaques. The phase II trial of lecanemab used florbetapir PET for amyloid imaging, and the reduction in amyloid signal observed may be, in part, attributable to a reduction in protofibrils.19
Lecanemab’s development history includes an informative phase I study and an innovative phase II trial. The agent has progressed to phase III and is in a regulatory environment that has viewed similar amyloid-lowering agents favourably. The biological target and mechanism of action of lecanemab are genetically linked; lecanemab has a biomarker that confirms the presence of AD (shown on amyloid positron emission tomography [PET]), a biomarker that demonstrates target engagement (shown on amyloid PET), safety biomarkers to ensure its safe use (shown on MRI) and preliminary support for clinical benefit.5,17,19 Innovative aspects of the development programme include the staggered parallel design of the phase I studies, inclusion of an early AD population, use of a biomarker to confirm the diagnosis of AD, implementation of a target engagement biomarker, adoption of the ADCOMS as an outcome, and application of a Bayesian adaptive dose-finding strategy. These factors provide the foundation for lecanemab advancing to phase III trials and possible successful development as a therapy for early AD.13,14,27











