Friedreich’s ataxia (FRDA), a neurodevelopmental and progressive neurodegenerative disease, is the most common inherited form of ataxia, with disease incidence as high as 1 in 29,000 in Caucasian populations.1 Patients typically present with ataxia from ages 7 to 15 years and lose the ability to walk by their mid-twenties.2 Other common features include cardiomyopathy, diabetes, spasticity, hearing and vision loss, and scoliosis.2 The primary cause of death is cardiomyopathy, with an average age of death of 37.5 years.2
FRDA results from mutations in the frataxin (FXN) gene that ultimately reduces the amount of functional frataxin protein in the cell. This is an autosomal recessive disease typically caused by a biallelic trinucleotide guanine-adenine-adenine (GAA) repeat expansion.3–6 In around 4% of cases, however, there is a repeat expansion in combination with a point mutation or large deletion on the paired allele.4–6
This article discusses the impending use of omaveloxolone in the treatment of FRDA, which has sparked much excitement since its US Food and Drug Administration (FDA) approval.7–9 It primarily aims to review the underlying cellular pathology and proposed mechanism of omaveloxolone in FRDA. Furthermore, the MOXIe study (RTA 408 capsules in patients with Friedreich’s ataxia; ClinicalTrials.gov identifier: NCT02255435), the multipart clinical trial that provided the support for the FDA approval of omaveloxolone in FRDA, is discussed in detail.2 This discussion will describe the challenges faced in clinical trials in FRDA and rare diseases more broadly. Finally, other therapies under investigation are briefly reviewed.
The cellular pathology of Friedreich’s ataxia
The basic pathological mechanism underlying FRDA reflects its decreased FXN expression. This decreased expression leads to two interrelated mechanisms of cell death: defective mitochondrial function and increased susceptibility to oxidative stress. FXN is a mitochondrial protein important for the generation of iron–sulfur clusters,10,11 prosthetic groups that are important to the function of the electron transport chain and a variety of other cellular enzymes.12,13 As such, the absence of frataxin leads to oxidative stress and mitochondrial dysfunction. This is further exacerbated by altered nuclear factor (erythroid-derived 2)-like 2 (Nrf2) signalling in FRDA. Nrf2 is a transcription factor that activates the expression of antioxidant genes, decreases the expression of pro-inflammatory genes, and helps generate co-substrates for mitochondrial adenosine triphosphate production.14,15 In the normal cellular state, oxidative stress causes Nrf2 translocation to the nucleus to increase the expression of antioxidant genes, protecting cells from damage. In FRDA, Nrf2 fails to translocate to the nucleus upon induction of oxidative stress, leading to the downregulation of Nrf2 targets.16 As a result, the ability of the cell to handle baseline oxidative stress is impaired; this is exacerbated by impaired energy production (Figure 1).
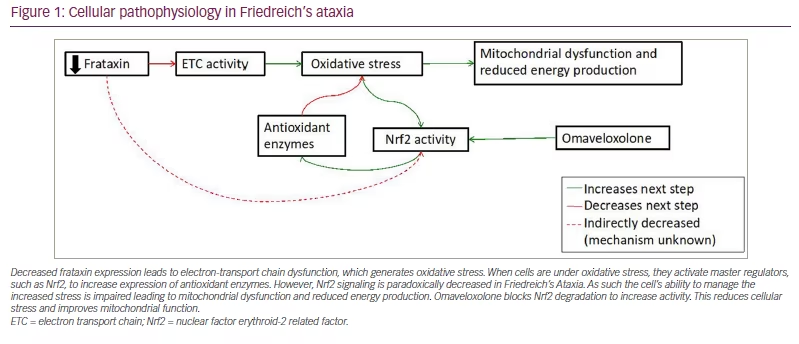
Decreased frataxin expression leads to electron-transport chain dysfunction, which generates oxidative stress. When cells are under oxidative stress, they activate master regulators, such as Nrf2, to increase expression of antioxidant enzymes. However, Nrf2 signaling is paradoxically decreased in Friedreich’s Ataxia. As such the cell’s ability to manage the increased stress is impaired leading to mitochondrial dysfunction and reduced energy production. Omaveloxolone blocks Nrf2 degradation to increase activity. This reduces cellular stress and improves mitochondrial function.
ETC = electron transport chain; Nrf2 = nuclear factor erythroid-2 related factor.
This mechanism suggests Nrf2 as a potential treatment target in FRDA. Nrf2 can be activated through exercise,17 calorie restriction,18,19 and natural and chemical activators.20 The efficacy and downstream functional consequences of each of these exercises depend on specific protein–protein interactions and vary widely according to the method of activation and dose used. The use of Nrf2 activators has been explored in multiple diseases: dimethyl fumarate is used in both multiple sclerosis and psoriasis,21,22 JM17 for spinocerebellar ataxia type 3,23 and RTA 402 for a variety of malignancies and chronic kidney disease.24–27
Omaveloxolone is a second-generation semisynthetic oleanane triterpenoid compound that binds to adapter protein Kelch-like ECH associated protein 1 to decrease ubiquitination of Nrf2,11 thus increasing cellular Nrf2 levels and improving both the cell’s ability to handle oxidative stress and produce energy. In vitro models have shown that omaveloxolone restores mitochondrial function in fibroblasts from both mouse models of FRDA and from patients with FRDA.11 The MOXIe study evaluated the clinical effect of omaveloxolone and will be discussed in detail below.
Recent clinical trials of agents targeting Friedreich’s ataxia
In addition to omaveloxolone, multiple agents have been considered for the treatment of FRDA. Clinical trials for these drugs have used a variety of outcome measures and shown variable efficacy.
A0001 (Edison Pharmaceuticals, Mountain View, CA, USA) is thought to improve mitochondrial function as an analogue of coenzyme Q10 and idebenone with greater bioavailability.28 A double-blind, randomized, placebo-controlled phase II trial (ClinicalTrials.gov identifier: NCT01035671) of this agent showed no significant change in the primary outcome measure, disposition index, a measure of diabetic tendency.28 However, the Friedreich’s Ataxia Rating Scale (FARS) score, a validated neurological exam-based scale used to provide a quantifiable measure of neurological function in patients with FRDA, significantly improved at both trial doses compared with placebo.11,29
A phase IIb study (ClinicalTrials.gov identifier: NCT01728064) investigating an almost identical molecule to A0001 that augments synthesis of the antioxidant glutathione, vatiquinone (PTC-743 [previously known as EPI-743] PTC Therapeutics, South Plainfield, NJ, USA), used low-contrast visual acuity as the primary outcome measure. The primary outcome did not reach statistical significance.30 However, a comparison with natural history data suggested a possible benefit.30 Vatiquinone is being evaluated in on-going trials (ClinicalTrials.gov identifiers: NCT05485987 and NCT04577352) in largely paediatric cohorts. One of these cohorts recently reported somewhat disappointing results. The trial failed to reach its primary endpoint, it saw improvement in the modified Friedreich’s Ataxia Rating Scale (mFARS), but showed a modest improvement in the upright stability scale and a significant improvement in fatigue.31 It is unclear whether this represents a path forward.32
Interferon gamma-1b (ACTIMMUNE®, Horizon Therapeutics PLC., Dublin, Ireland), a cytokine involved in the immune response and iron metabolism, significantly upregulates expression of FXN in multiple cell types from patients with FRDA and mouse models.33 It increases blood frataxin levels in a subset of patients with FRDA in a dose-escalation, phase II study (EudraCT2012-001881-14)34; however, these results were not statistically significant. There was also no significant change in score in the Scale for Assessment and Rating of Ataxia, a quantitative neurological exam score for assessing the severity of ataxia. A separate study of adolescents demonstrated a five point decrease in the FARS score, possibly indicating greater efficacy in a younger age group less affected by neurodegeneration.35 36 However, a phase III trial of interferon gamma-1b in subjects aged between 10 and 25 years failed to show a significant improvement in the primary outcome measure, the mFARS score, and changes in blood or buccal frataxin levels.37
Epicatechin ([+]-EPI, Epirium Bio, Inc., San Diego, CA, USA) is a natural flavonoid that helps regulate cellular antioxidant responses via the Nrf2 pathway.38 This molecule has been studied in a phase II study of paediatric patients with FRDA. While there was significant improvement in cardiac-related outcomes, primary neurologic outcomes did not show statistically significant improvement.38
Finally, RT001 (Retrotope, Inc., Los Altos, CA, USA) is a compound thought to reduce cellular damage and improve mitochondrial function through the inhibition of lipid peroxidation. A double-blind, placebo-controlled trial (ClinicalTrials.gov identifier: NCT02445794) showed this compound to be well tolerated and improved peak workload in FRDA.39 However, RT001 failed to show benefit in a further double-blind, placebo-controlled trial.40
Some agents, such as A0001 and epicatechin, initially showed promising results but failed to move forward, largely for pragmatic reasons or due to the relative similarity of such agents to other agents related to antioxidants. At present, a novel group of therapies designed to restore frataxin are being evaluated in clinical trials. Gene therapy trials are open for cardiac disease in FRDA (ClinicalTrials.gov identifiers: NCT05445323 and NCT05302271), and CTI-1601 (ClinicalTrials.gov identifier: NCT05579691) (formerly known as trans-activator of transcription [TAT]-frataxin; Larimar Therapeutics, Bala Cynwyd, PA, USA) has shown promising results in restoring frataxin levels in peripheral tissue (LARIMAR study).41 Finally, bypassing the silencing induced by the GAA1 repeat may also be promising.42 Thus, a variety of approaches remain.
The highly variable clinical presentation combined with a slow rate of change over time and the rare incidence of disease make the design and interpretation of rare disease clinical trials especially challenging.43,44 Research in FRDA is uniquely supported by two large natural history studies that have generated a better understanding of the disease presentation and variability, progression over time, and measurement strategies.29,45–48 This is useful to determine subgroups and expected responses when designing clinical trials, and provides a comparison population.
Despite this advantage, many clinical trials in FRDA have been limited by factors such as insensitive outcome measures, short duration, suboptimal target patient population and incorrect dosing.44 For example, when advancing from smaller to larger studies with a more heterogenous population of patients, clinical rating scales may perform differently, and a longer study duration may be needed to detect change. Clinical outcomes measures such as the FARS score, the timed 25-foot walk and the nine-hole peg test may not be able to detect small improvements in neurological functions that occur over the time course of brief trials lasting only a few months. In addition, small sample sizes make it difficult to identify responders versus non-responders and further define characteristics that may contribute to the response.
Another challenge with FRDA, like many rare diseases, is that the pathophysiology is not well understood. While it is known that FRDA is a progressive disease, this may be superimposed on the preclinical loss-of-function, and baseline characteristics that predict disease severity, such as age of onset or GAA1 repeat length, are likely not yet defined.44 Furthermore, some changes may be related to modifiable events, such as metabolic dysfunction, while others are secondary to irreversible events, such as neuronal loss, that are not likely to be amenable to pharmacologic intervention. There are also a few useful biochemical biomarkers identified in FRDA. Frataxin is only a valid biomarker in trials where the mechanism of action is the upregulation of FXN expression.43 Furthermore, determining the optimal and minimum effective dose is critical in evaluating the efficacy of an agent, and phase II trials are the best time to do this. The trials for the therapies described above (A0001, vatiquinone, interferon gamma-1b and RT001) generally used only two doses plus placebo. Moreover, the phase I/II studies investigating RT001 (n=19) and interferon gamma-1b (n=9) had a small sample size; thus, the data of these two studies could not define a detailed dose–response relationship, perhaps explaining why the phase III studies were not successful.
The success of omaveloxolone is due in part to how the trials were designed to overcome these challenges. The MOXIe trial was not only one of the largest clinical trials conducted in FRDA, but also one of the longest due to the incorporation of the extension phase. Additionally, it focused on a well-defined target population to limit variability and started with a dose-optimization phase, including eight different doses.
The MOXIe study
The MOXIe study is a multi-part, placebo-controlled, phase II study evaluating the use of omavaloxolone in FRDA designed to mitigate many of the above challenges (Figure 2).1,23 The study was divided into two parts: Part 1, which was 12 weeks and focused on determining the optimal dose; and Part 2, which evaluated the effect of omavaloxolone on neurological function after 48 weeks of treatment.2,49 These parts were then followed by an open-label extension phase to determine long-term safety, the durability of response and the relative impact of early initiation of treatment.50 Participants were between 16 and 40 years old. Baseline function was characterized by the mFARS score. There are four components to the score: bulbar, upper limb coordination, lower limb coordination and upright stability. Total scores range from zero to 99, with lower scores indicating better neurologic function. Subjects were tested by the lead investigator at each study site and the examiners performing neurological assessments were blinded to all other results, including the laboratory values. Patients were excluded if they had uncontrolled diabetes, clinically significant cardiac disease (low ejection fraction or known unstable arrhythmia), significant laboratory abnormalities or interfering medical conditions.2
Part 1
Part 1 was a placebo-controlled, dose-ranging study aimed at addressing drug safety and tolerability.49 A total of 69 patients took omavaloxolone orally for 12 weeks at doses ranging from 2.5 mg to 300.0 mg per day. This allowed identification of both the minimal and the maximal effective dose. Overall, the drug was well tolerated. Optimal pharmacological responses were observed at 80 mg and 160 mg. There was no improvement in the primary outcome, peak workload in maximal exercise testing, at any dose; however, the 160 mg dose did improve the secondary outcome, change in the mFARS score, compared with the baseline and placebo.
Part 2
Part 2 enrolled 103 individuals across 11 study sites in the USA, Europe and Australia in a randomized, placebo-controlled clinical trial.2 Subjects were between 16 and 40 years of age (inclusive), had a mFARS score ranging between 20 and 80 at baseline and could ride an exercise bike against no resistance for 3 min. This created a cohort of moderately affected subjects who were still largely ambulatory. Patients either received 150 mg of omavaloxolone or placebo per day for 48 weeks. Randomization was stratified according to pes cavus presence based on Part 1, which suggested lower response among those patients;49 the presence of pes cavus may reflect a more neurodevelopmental, rather than neurodegenerative, phenotype. Data were collected at screening and at Weeks 4, 12, 18, 24, 36 and 48. There was an additional follow-up safety visit at Week 52, 4 weeks after the last dose. A total of 94 patients (91%) completed treatment through to Week 48, including 44 (86%) of 51 participants randomized to omaveloxolone and 50 (96%) of 52 randomized to placebo. The primary outcome measure was a change in mFARS score from baseline. Secondary outcome measures included Patient Global Impression of Change, Clinicial Global Impression of Change, nine-hole peg test, timed 25-foot walk, Activities of Daily Living score, frequency of falls and peak work during maximal exercise testing.2
Overall, treatment with omaveloxolone significantly improved neurological function. Patients who received omaveloxolone overall had lower mFARS scores at Week 48 by 2.40 ± 0.96 points compared with those who received placebo (p=0.014; n=82). The magnitude of the observed improvements is equivalent to approximately 2 years of FRDA progression, as judged by age-matched cohorts in a natural history study, Friedreich Ataxia Clinical Outcome Measures Study.2,51 This considerable improvement can rationally be viewed as meaningful, given that almost all patients with FRDA continue to have disease progression yearly. In addition, single items of the mFARS can generally be matched to specific clinical tasks, such that a one point improvement in mFARS score may present better function on a specific task that is clinically meaningful. Nonetheless, the exact minimum change in mFARS score to be clinically meaningful remains a topic of discussion. Although treated patients showed improvement in each of the four components within the mFARS assessment relative to placebo, the greatest effect was seen with upright stability. The relative improvement in the mFARS scores persisted when the study population was stratified by age and sex, with the greatest improvements in mFARS witnessed in patients younger than 18 years. Paediatric patients (ages 16 and 17) treated with omaveloxolone improved by -1.63 ± 1.78 points at Week 48, whereas patients who received placebo worsened by 2.52 ± 1.18 points, resulting in a placebo-corrected improvement of -4.16 ± 2.15 points (p=0.057).
With regard to secondary outcome measures, mean Patient Global Impression of Change and Clinician Global Impression of Change scores at Week 48 numerically improved in treated patients, but did not statistically differ between treatment groups (p=0.13). In addition, withdrawal of the active agent at Week 48 was associated with a return toward placebo values, consistent with only some of the benefit being endured at Week 48; however, placebo values were not reached. Patients who received omaveloxolone experienced headache, nausea, fatigue and transient reversible increases in aminotransferase levels without increases in total bilirubin or other signs of liver injury. Such changes in aminotransferase levels were maximal within the first 12 weeks of treatment and trended back toward baseline as therapy continued.2
Extension phase
The open-label extension phase compared the difference in the mFARS score at the end of the 48 weeks placebo-controlled period (Part 2) with a follow-up of up to 124 weeks. Patients who completed MOXIe Part 2 were eligible to enrol in the open-label extension.50 The 39 subjects who were originally randomized to placebo in Part 2 initiated treatment with omaveloxolone in the open-label phase. This allowed for a delayed-start analysis comparing the difference in mFARS score between those who started omaveloxolone at Week 0 (omaveloxolone–omaveloxolone group, n=34) and those who started at Week 52 (placebo–omaveloxolone group, n=39). All patients received 150 mg of omaveloxolone one time daily. The mFARS scores were completed on Day 1 and every 24 weeks for a total of 72 weeks. Both subjects and examiners were blinded to the original treatment group from Part 2. Due to interruptions to on-site visits caused by the coronavirus disease 2019 pandemic, multiple mFARS values are missing at Week 48 and 72.
The omaveloxolone–omaveloxolone group had a slightly more advanced disease compared with the placebo–omaveloxolone group as measured by three baseline characteristics, each associated with a more advanced disease: higher average baseline of the mFARS scores (40.9 compared with 38.8 in the placebo–omaveloxolone group); longer average GAA1 repeat length (associated with worse disease severity; 739.2 compared with 693.8 in the placebo–omaveloxolone group); and a greater proportion of individuals with cardiomyopathy (19 compared with 12 in the placebo–omaveloxolone group). Ten patients discontinued treatment during the extension phase, eight in the placebo–omaveloxolone group and two in the omaveloxolone–omaveloxolone group. The difference in the mFARS score between omaveloxolone and placebo observed at the end of the placebo-controlled period (mean difference -2.17 ± 1.09) was preserved at the end of the delayed-start period (mean difference -2.91 ± -1.44).50
Both groups also showed persistent benefit compared with natural history control data external to the MOXIe study, with slower disease progression (placebo–omaveloxolone group: 56%; omaveloxolone–omaveloxolone group: 61%) over 3 years.51 Serious adverse events were reported in 13 (8.7%) patients, 8 (7.5%) of which were in the placebo–omaveloxolone group.24 All of the serious adverse events were considered by investigators to be unrelated to the study drug. Overall, the delayed start analysis showed a persistent benefit of omaveloxolone on disease course, and the sustained benefit shown in the omaveloxolone–omaveloxolone group could not be recovered by individuals initially randomized to placebo, implying the benefit of starting omaveloxolone treatment earlier.50
Other therapies under investigation
Multiple other therapies are currently under investigation for the treatment of FRDA, including drugs with a variety of different mechanisms, such as antioxidants, regulators of gene expression and gene therapy. Antioxidants could help mitigate mitochondrial oxidative stress from decreased frataxin levels; however, to this point, simple antioxidant activity has shown little benefit. Vatiquinone is currently being investigated in the phase II/III study MOVE-FA (ClinicalTrials.gov identifier: NCT04577352), which is evaluating its safety and efficacy in patients aged seven and older,52 and a phase II trial (ClinicalTrials.gov identifier: NCT05485987), which is assessing its pharmacokinetics in children younger than seven.53,54 These studies propose vatiquinone as a selective ferroptosis inhibitor rather than a non-specific antioxidant. Two other antioxidant-related agents under investigation are epicatechin and peroxisome proliferator-activated receptor-γ agonists (e.g. pioglitazone and letiriglitozone). Peroxisome proliferator-activated receptor-γ agonists act as antidiabetic agents by augmenting insulin sensitivity and normalizing serum glucose levels;55 however, they also regulate numerous cellular functions, such as mitochondrial biogenesis and reactive oxidative species metabolism.53,56
Frataxin restoration remains the definitive approach for treating FRDA. Regulators of FXN gene silencing are one approach for frataxin restoration. Histone deacetylase (HDAC) inhibitors disrupt gene silencing by increasing the histone acetylation of FXN and promoting increased frataxin.57 Several HDAC inhibitors, including 2-aminobenzamide class I HDAC inhibitors, upregulate FXN gene expression in FRDA pluripotent stem cells and FRDA lymphocytes.58,59 Further research on HDAC inhibitors has been limited by adverse side effects; however, synthetic transcription factors (e.g. Syn-TEF1) may enable the transcription of FXN across trinucleotide GAA repeats responsible for undermining frataxin expression.60 Frataxin protein attached to the TAT sequence can deliver frataxin into the mitochondrial matrix. In this approach, the TAT peptide allows endocytosis of the TAT-frataxin molecule while the mitochondrial targeting sequence endogenous to frataxin facilitates transport into the mitochondrial matrix as a protein replacement therapy.61 A phase I clinical trial (ClinicalTrials.gov identifier: NCT04176991) was conducted in 2020 to evaluate the safety and efficacy of CTI-1601 (TAT-frataxin) in the treatment of FRDA; preliminary reports on this trial suggest that CTI-1601 can restore frataxin levels.41
Lastly, gene therapy constructs are under investigation for treating FRDA. Adeno-associated virus vectors carrying human frataxin prevented cardiomyopathy, reversed cardiomyopathy following heart failure and reversed sensory neuropathy in mouse models of FRDA.62,63 These preclinical data are particularly exciting, given that cardiomyopathy and heart-related dysfunction are the main causes of death in patients with FRDA.53 There are two active clinical trials using this construct (ClinicalTrials.gov identifiers: NCT05445323 and NCT05302271).64 There is also interest in using antisense oligonucleotides complementary to the trinucleotide repeats in FXN to increase gene expression, but the data have not yet shown utility.65 Similarly, a recent phase I clinical trial (ClinicalTrials.gov identifier: NCT05285540) investigated a small molecule, DT-216, designed to restore transcription and production of natural frataxin, results are not yet available from this study.42
Conclusions
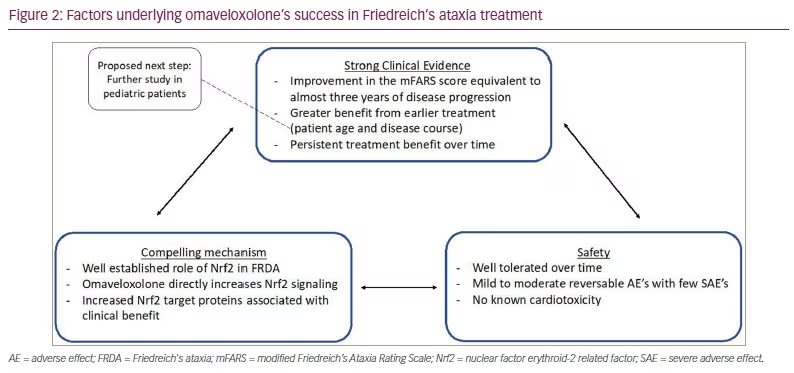
The FDA approved omaveloxolone for FRDA in early 2023,66 the first treatment available to patients with FRDA in the USA. Although different sites worldwide participated in the MOXIe study, approval has not yet been granted outside of the USA. This major step is the result of a compelling cellular mechanism combined with strong clinical evidence and safety data from well-designed clinical trials. The MOXIe study was carefully designed to address many of the difficulties faced by rare disease clinical trials, including prior FRDA studies. From the point of view of basic science, omaveloxolone is supported by substantial cellular data; however, there is little to no evidence from mouse models of FRDA, in contrast to many drugs in development. Nonetheless, this shows that, although understanding the underlying mechanism of a drug is important, animal data are not necessarily required.
The study employed rigorous inclusion and exclusion criteria to target a more homogeneous study population. This helped to address the inherent variability in clinical presentation and disease course in rare diseases and enriched the population for features likely to respond to the drug. Although this strategy may help to identify clinical patients most likely to respond to the drug, it does limit generalizability. For example, children under 16 years and those with lower exercise tolerance (including lower motor ability and reduced cardiac function) were not included in the study; therefore, it is unclear how these populations may benefit from omaveloxolone treatment. As no safety data are available on patients younger than 16, the FDA approval excluded this patient population. A key aspect of the approval granted for omaveloxolone is the absence of major safety events, this allowed for greater flexibility in attaining a favorable risk–benefit ratio despite imperfections in the data showing benefit.
The MOXIe study was also unique in that it included a broad dose range, which allowed to develop a well-mapped dose-response curve. Additionally, the long follow-up time increased sensitivity to smaller clinical changes, and comparison with natural history data showed how the drug alters the natural disease course.
Overall, the success of MOXIe can be summarized in a few concepts:
-
Well-defined mechanism
-
Relatively safe drug
-
Long follow-up to demonstrate endurance
-
Optimized dose and subject population
-
Detailed analysis with multiple statistical paradigms demonstrating consistent results
We are hopeful that omaveloxolone will have a promising impact for patients with FRDA, and we look forward to see how this landmark will support progress in other therapies under investigation.