Cerebral amyloid angiopathy (CAA), also known as congophilic angiopathy, is a recognized cause of lobar intracerebral haemorrhage in patients above the age of 50 years.1 It is known to be due to deposition of a variant of amyloid (beta-amyloid plaque) in cerebral blood vessels. Beta-amyloid plaque is formed by the degradation of amyloid precursor protein via secretases.2 The plaque is then deposited within the cerebral vasculature, primarily the leptomeningeal and cortical vessels. Initially, the amyloid-beta plaque is deposited in the tunica media and adventitia of blood vessels and impairs smooth muscle cells,3 leading to fibrinoid necrosis. Patients who have CAA are at significantly increased risk for intracerebral haemorrhage, especially those who are anticoagulated.4
CAA-related inflammation (CAA-ri) encompasses a subset of patients with CAA who present with seizures, subacute cognitive decline, vasogenic oedema and microbleeds on magnetic resonance imaging (MRI), vasculitis and perivasculitis.5 Here, we present a case of CAA-ri in a rural area with an initial presentation of diffuse encephalopathy rather than the classic lobar haemorrhage.
Case presentation
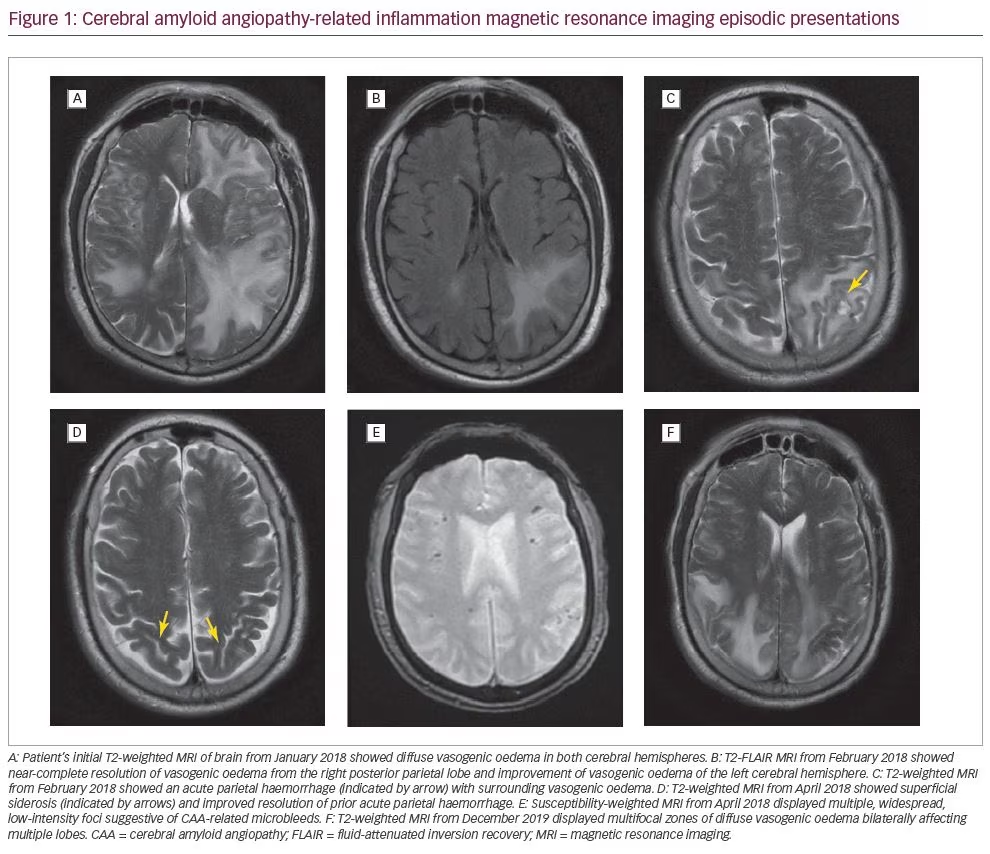
In January 2018, a 71-year-old Caucasian male with a history of atrial fibrillation and hypertension presented with acute confusion and a 2-week history of right greater than left sided arm and leg weakness with ataxia. He was subsequently hospitalized at a rural hospital with an initial diagnosis of cerebral vasculitis based on computed tomography (CT) imaging showing bilateral vasogenic oedema, a 4 mm midline shift and a right thalamic lacunar infarct. Magnetic resonance imaging (MRI) showed diffuse vasogenic oedema in both cerebral hemispheres, intervening zones of gyral oedema, a tiny acute petechial haemorrhage and leptomeningeal enhancement with diffuse signal dropout supportive of amyloid angiopathy (Figure 1A). T2 gradient echo (GRE) findings included innumerable findings of signal dropout scattered throughout the right and left cerebral hemispheres peripherally at zones of grey–white matter differentiation. GRE findings also included signal dropout within the gyral cortex, subsequently surrounded by vasogenic oedema. This pattern is highly suggestive of amyloid angiopathy. Initial supportive care at the rural hospital included intravenous dexamethasone before he was subsequently transferred to a metropolitan neurocritical care unit 150 miles away. During the process of narrowing down the differential diagnoses, he was treated with high-dose dexamethasone, acyclovir, azathioprine and levetiracetam in the event he was suffering from central nervous system (CNS) vasculitis, infectious encephalitis or autoimmune encephalitis. Subsequent laboratory work, including a lumbar puncture, ruled out herpes simplex virus, tuberculosis, syphilis, lupus and fungal causes of encephalitis or meningitis. Autoimmunity studies were also negative. He remained on dexamethasone for his vasogenic oedema, and azathioprine was continued for treatment of possible CNS vasculitis, which could not be ruled out by the lab tests.
Magnetic resonance angiogram (MRA) also revealed severe left vertebral artery stenosis. The patient’s hospital course was complicated by development of a pulmonary embolism and deep vein thrombosis. An inferior vena cava filter was placed. At the time, his diagnosis remained unclear. The patient was stabilized, continued on azathioprine and discharged.
A follow-up MRI in February 2018 (Figure 1B) showed near-complete resolution of vasogenic oedema from the right posterior parietal lobe and improvement of vasogenic oedema of the left cerebral hemisphere more than right cerebral hemisphere. However, this MRI also showed an acute haemorrhage in the left posterior parietal lobe (0.9 cm x 1.4 cm) with inflammation (Figure 1C). The patient followed up with his primary care physician who prescribed an additional course of dexamethasone for the vasogenic oedema. The patient also began physical therapy and occupational therapy as recommended by a vascular neurology clinic.
In March 2018, the patient followed up with his rural neurologist who continued azathioprine for possible vasculitis. A repeat MRI in April 2018 (Figure 1D) showed resolution of vasogenic oedema, resolution of the acute haemorrhage in the parietal lobe, and superficial siderosis suggestive of amyloid angiopathy. The GRE sequence again demonstrated extensive blooming artifact within the peripheral aspect of the brain often seen at cortical grey–white matter junctions; a pattern typical for amyloid angiopathy.
In July 2018, the patient’s MRI showed patchy haemosiderin and chronic stable enlargement of the pituitary gland. The patient was considered safe to start aspirin and clopidogrel in September 2018 indicated by his history of severe left vertebral artery stenosis found during his hospital course in January. Rivaroxaban was not started due to risk of bleeding, despite history of atrial fibrillation. A repeat MRA, performed in November 2018, was negative other than severe left vertebral artery stenosis. The patient was stable on his regimen of medications, with no significant changes in follow-up imaging or clinical evaluation until December 2019, when he presented to the hospital with stroke-like symptoms. A CT brain showed severe hypoattenuation within bilateral parietal lobes and left posterior frontal subcortical white matter. Brain MRI (Figure 1F) displayed multifocal zones of diffuse vasogenic oedema bilaterally affecting multiple lobes. Gyral oedema was present in bilateral parietal lobes and the left frontal lobe, along with widespread foci of micro-haemosiderin through bilateral cerebral hemispheres. GRE sequence demonstrated widespread foci of signal dropout within the peripheral margin of the brain at cortical grey–white matter interfaces involving virtually all lobes of the brain, suggesting regions of micro-haemosiderin deposition, favoured to be secondary to cerebral amyloid disease. After a similar stabilization as before, the patient now resides at a nursing and rehabilitation centre, where he completed a dexamethasone taper for vasogenic oedema.
Discussion
This case of an elderly patient with CAA represents a subset of patients that present with CAA-ri. The typical presentation of CAA follows a spontaneous lobar haemorrhagic pattern; however, some patients may present with seizures, subacute cognitive decline or headaches, and show asymmetric vasogenic oedema, microbleeds on MRI and vasculitis or perivasculitis on neuropathologic examination.5
In our case, we observed a patient who initially presented with acute confusion and stroke-like symptoms, and was found to have bilateral vasogenic oedema. While CAA can be definitively diagnosed with biopsy, imaging findings of haemorrhage are characteristic. A brain biopsy was not obtained in this case due to limited resources in a rural setting. The initial imaging findings in our patient showed widespread vasogenic oedema with no lobar haemorrhages. His first MRI showed only an acute tiny petechial haemorrhage; however, this was resolved on a follow-up MRI when the patient was asymptomatic after improvement from the initial episode. His next episode also showed vasogenic oedema with no haemorrhages. A key feature for CAA diagnosis in our patient was superficial siderosis and widespread, low-intensity foci suggestive of CAA-related microbleeds found on imaging.
A similar presentation to that discussed here has been reported in the literature, whereby a middle-aged male presented with one generalized tonic-clonic seizure.6 He underwent subsequent MRI over time and was found to have CAA-ri presenting as non-haemorrhagic diffuse encephalopathy. In our case, due to multiple MRI reports revealing widespread, low-intensity foci suggestive of CAA, haemosiderin deposits noted by multiple radiologists, as well as repetitive episodes of inflammation, CAA-ri was the presumed diagnosis after ruling out the other prior-mentioned differentials. The diagnosis of CAA was also supported by the modified Boston Criteria, as this patient was above the age of 55 years with superficial siderosis and cortical bleeds.7
Our presentation is important in realizing the different imaging presentations that CAA can take in the form of CAA-ri. It is important not to delay treatment while waiting for laboratory results to return. Dexamethasone and azathioprine should be started immediately, as CAA and cerebral vasculitis should be considered possible diagnoses. It is also important to recognize that even though imaging may not reveal any initial lobar haemorrhage, patients with non-haemorrhagic presentation of CAA are still predisposed to haemorrhages due to amyloid weakening of blood vessels, as seen in this case. This pathologic process makes management of other comorbid conditions, such as hypertension, important.
Concluding remarks
CAA most commonly presents as a lobar intracerebral haemorrhage. The disease process of plaque deposition leads to fibrinoid necrosis and eventual haemorrhagic events. A subset of patients with CAA may present with CAA-ri. Presentations of CAA-ri may include episodes of vasogenic oedema and microbleeds on MRI, as seen in our patient. It is key to avoid premature exclusion of other pathologies, and begin prompt treatment of the vasogenic oedema with steroids and immunosuppressants until further work-up is complete.
Minor post-publication correction: Compliance with ethics statement updated