Introduction
Fabry disease is a treatable X-linked disorder caused by a deficiency in alpha-galactosidase. Decrease of the enzyme activity leads to accumulation of globosyltriaosylceramide (Gb3) in tissues resulting in progressive organ dysfunction especially of the heart, the renal system and the vascular system.
Frequency of Fabry Disease
The incidence of Fabry disease varies widely from setting to setting. In a recent neonatal screening study, a frequency of 1:4,600 live births was found.1 Other data suggest an incidence of 1:117000.2
Introduction
Fabry disease is a treatable X-linked disorder caused by a deficiency in alpha-galactosidase. Decrease of the enzyme activity leads to accumulation of globosyltriaosylceramide (Gb3) in tissues resulting in progressive organ dysfunction especially of the heart, the renal system and the vascular system.
Frequency of Fabry Disease
The incidence of Fabry disease varies widely from setting to setting. In a recent neonatal screening study, a frequency of 1:4,600 live births was found.1 Other data suggest an incidence of 1:117000.2
Females generally have a more heterogeneous clinical picture due to the variable enzyme activity within the different tissues (lyonisation effect).3 The disease is panethnic.
Pathogenesis
Decrease of alpha-galactosidase activity inhibits the formation of lactosylceramide, an essential step in the formation of ceramide from globoside within the lysosome.
Accumulation of Gb3 damages renal epithelium and respiratory epithelium, as well as myocardial cells, dorsal root ganglia, neurons and vascular endothelium and smooth muscle cells. The various involvements within the different organ system and the varying clinical picture lead frequently to a delay in aetiologic diagnosis.4
Mutations
More than 200 mutations have been identified within the alpha-galactosidase gene.5 In 90% of affected families point mutations are identified within the seven exons of the alpha-galactosidase gene. Insertional mutations have also been described. Small deletions within the gene are well known but may cause difficulties with genetic diagnosis. The role of some intronic splice site mutations remains controversial.6
Clinical Features
Systemic Characteristics
Several clinical features point to a high likelihood of Fabry disease.7 Cornea verticillata, a whirl-like clouding of the cornea, best seen with slit-lamp examination, in the absence of the use of amiodarone or chloroquine, is very specific. Angiokeratomas, asymptomatic, punctiform papules, often occurring in clusters, are located in the genital area or periumbilical region and less commonly in the mucosal tissues. Angiokeratomas are infrequently found in other diseases.8
Less specific but relevant features for the disease include proteinuria, progressive renal failure, cardiomyopathy, ventricular hypertrophy, myocardial infarction and arrhythmia. These manifestations, together with cerebrovascular syndromes, are the principal causes of the progressive disability and mortality due to Fabry disease.9
Cerebrovascular Features
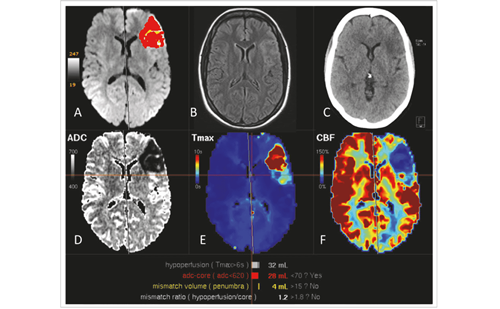
Typically, stroke is considered a manifestation of end-stage Fabry disease.10 However, in registries strokes occurring as early as childhood have been recognised.4 It is estimated that during the course of Fabry disease 16% of the patients will suffer a stroke. Strokes occur both in women and men.
The risk of stroke is increased 12-fold compared with age matched controls from the general population. It remains unexplained why they occur with a predilection for the posterior circulation.
In one series strokes occurred in the posterior circulation in up to 67% of Fabry disease patients.10 Stroke is typically ischaemic, but intracerebral hemorrhage has also been known to occur, maybe due to uncontrolled hypertension.11
Potential Stroke Mechanisms
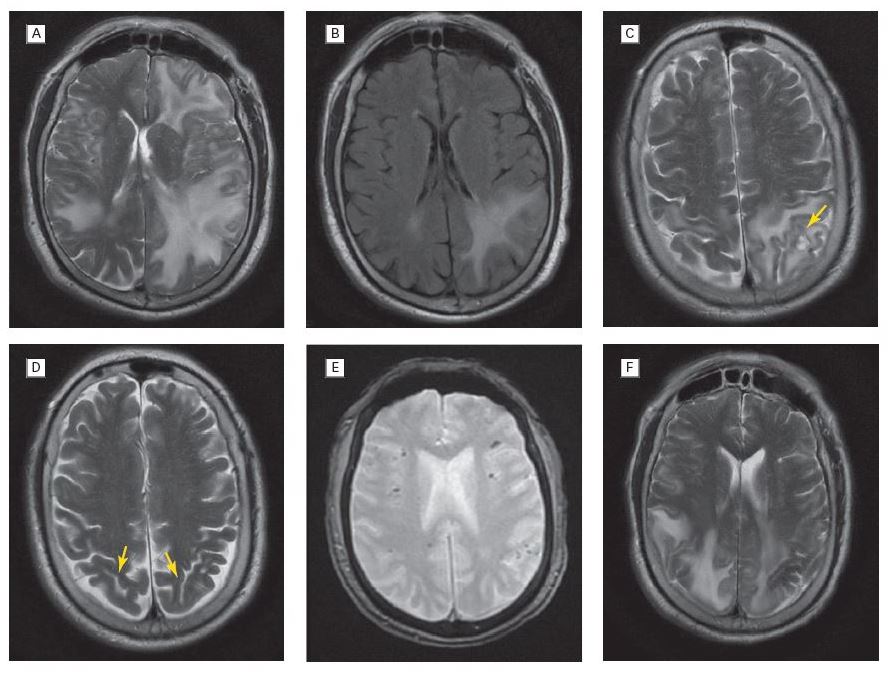
Several mechanisms can explain stroke in patients with Fabry disease. Hypertension due to renal dysfunction is the most important general risk factor for stroke and is associated with white matter disease, small vessel disease, intracerebral haemorrhage and endothelial dysfunction.11 Cardiomyopathy, valvular disorders and arrhythmias may cause cardiac emboli lodging in cerebral vessels. Apart from these consequences of systemic Fabry disease manifestations, Gb3 accumulation may lead to, albeit poorly understood, progressive vessel dysfunction, both from small vessels and from large vessels. White matter disease, a typical manifestation of small diameter vessels, occurs typically at a younger age than in patients without Fabry disease.3
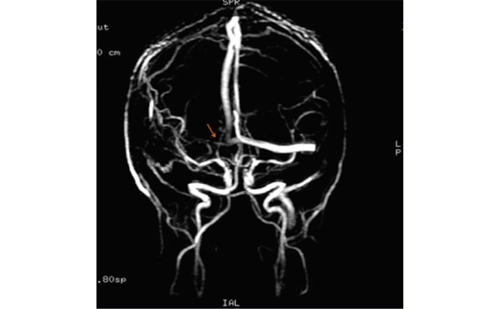
This may also lead to silent strokes and cognitive dysfunction. Sometimes patients are misdiagnosed with multiple sclerosis.11 Dolichoectasia, a typical large vessel disease, is frequently found. Emboli can form within the ectatic arteries, the artery may compress the brainstem and thrombus may occlude penetrating arteries of the brainstem. Chronic cerebral hyperperfusion, but also chronic hypoperfusion of white matter has been described.12-13 Whether Fabry disease, can lead to accelerated atherosclerosis has not been studied. Other mechanisms may also partly explain stroke in individual patients: for instance, a prothrombotic state may play a role.14-15
Diagnosis
Diagnosis can be reliably made in males using measurement of alpha-galactosidase enzyme activity with subsequent confirmation by genetic analysis. Pseudo-deficiency occurs very rarely.16 A definitive diagnosis in females can only be made using genetic analysis, as enzyme activity can be entirely normal in up to 20% of patients.
Involvement in Cryptogenic Stroke
Up to 30% of ischaemic strokes remain unexplained after thorough investigation. This is quite disconcerting for the patient faced with uncertainty about the correct diagnosis and prognosis. Cryptogenic stroke is more common in patients with stroke occurring at a young age, defined as age 55 or less. In a series of 721 cryptogenic young stroke patients from several centres in Germany, Fabry disease was diagnosed in 4.9% of males and in 2.4% of females. Strokes occurred mostly in the posterior circulation. Dolichoectasia was described in about one-third of patients.
Evidence Base for Enzyme Replacement Therapy
Phase II and III trials have been performed to show the beneficial effect of ERT on pain, renal dysfunction and other surrogate markers of the disease. These trials led to approval of ERT by the US Food and Drug Administration (FDA) and by European health authorities. Treatment is currently given life-long by infusions given every other week. Dose adjustments are typically not performed, but this probably should be studied further. A double-blind, placebo-controlled phase II trial of ERT included 26 male patients with neuropathic pain who received 0.2mg/kg every two weeks for 22 weeks.17 The primary efficacy end-point was pain with pain medication withdrawn before evaluations.
The neuropathic pain improved in 14 patients treated with ERT and changed little in the 12 placebo patients. Mean creatinine clearance did not change for patients receiving ERT but decreased by 16.1mL/min for placebo patients receiving placebo. Other parameters were also evaluated in these patients: the elevated regional cerebral blood flow and abnormal cerebrovascular responses, as measured by positron emission tomography and transcranial Doppler studies, were significantly reduced or improved after long-term follow-up.
A phase III multinational, multicentre, randomised, double-blind, placebo-controlled study evaluated the safety and effectiveness of ERT in 58 patients who received 1mg/kg of ERT or placebo every two weeks for 20 weeks.18 The primary efficacy end point was the percentage of patients whose renal capillary endothelial Gb3 deposits cleared to normal or near normal. Gb3 was cleared in 20 of 29 patients (69%) treated with ERT compared with 0 of 29 placebo patients receiving placebo from the renal capillary endothelium.
Both ERT and placebo groups had improvements in pain and quality-of-life assessments. However, patients in this study were not selected for pain and therapy with pain medications was not stopped during treatment.
Future Perspectives
The high frequency of Fabry disease among patients with cryptogenic stroke is surprising. This result is important since therapy can be provided and complications of Fabry can be managed using ERT in these patients. Several studies are now currently underway to estimate whether these results can be reproduced. These studies will examine patients with all types of stroke, not only patients with unexplained stroke, as strokes in Fabry disease can be associated with cardiac disease, small and large vessel stroke and even coagulopathy.
Although ERT is clearly beneficial for preservation of renal function and pain, it remains unknown whether ERT actually prevents recurrent stroke.
This presents a difficult question, that will probably never be satisfactorily resolved, given that placebo controlled trials are not considered ethical anymore with the current availabillty of ERT. ■