Neurologic manifestations in systemic sclerosis are rare, occurring in 0.8–5.6% of patients. Peripheral neuropathy is one of the most common neurologic abnormalities, with central nervous system involvement reported less frequently.1 Central nervous system (CNS) manifestation has been described in a few cases,1–3 and has been explained by a compressive mechanism or through metabolic disorders due to gastrointestinal tract involvement, or in relation to inflammatory diseases known to be associated with spinal cord involvement.1 We report a case of systemic sclerosis which presented as a multiple sclerosis (MS)-like condition.
Case report
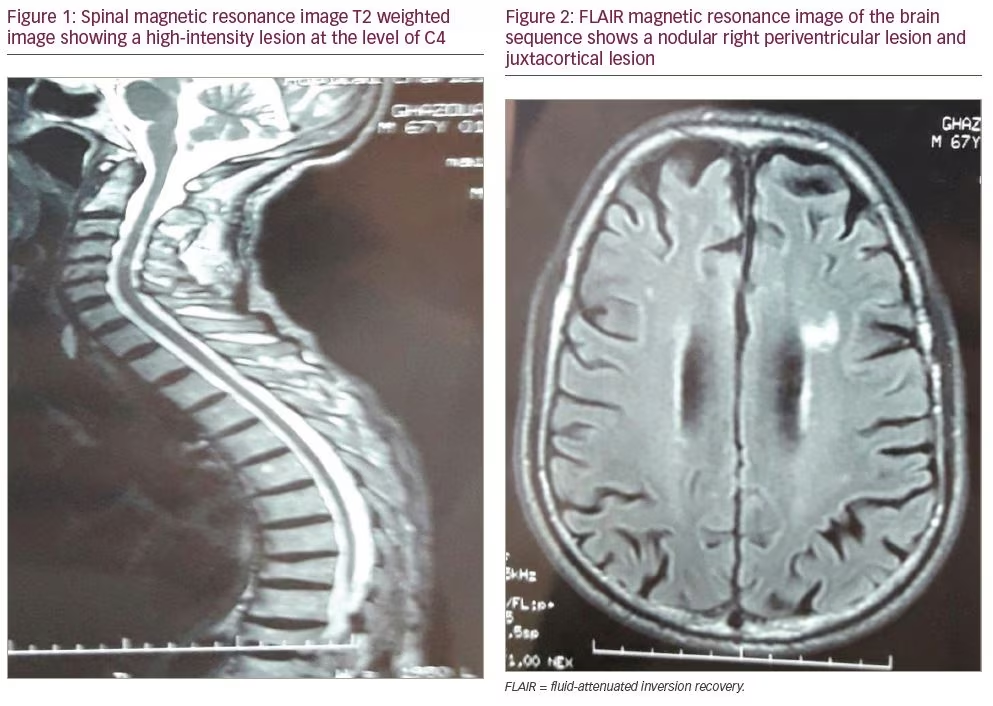
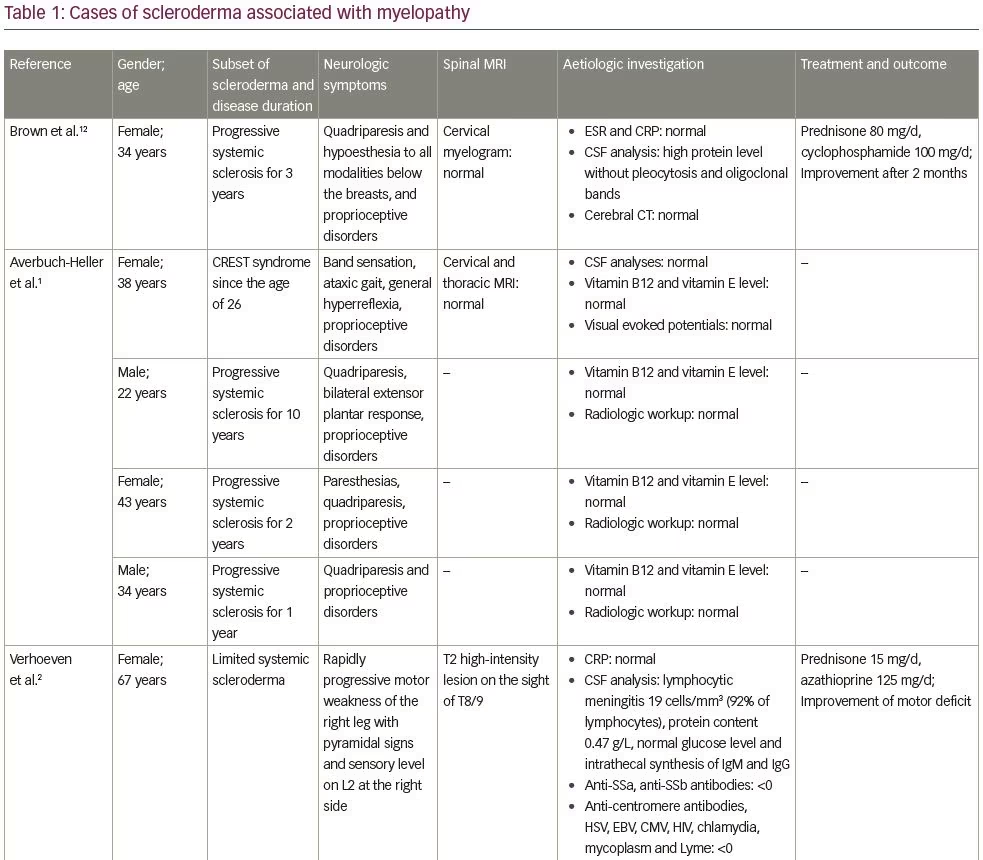
A 67-year-old man without any significant past medical history presented to our department with 2 years of progressive weakness in the lower limbs and urinary urgency. Neurologic exam found exaggerated deep tendon reflexes in all four limbs, mainly in the lower limbs, and spastic paraparesis. No motor deficit was found in upper limbs. Additionally, we did not find sensory or visual disorders or ataxia. Raynaud’s phenomenon and puffy fingers were noted. Spinal magnetic resonance imaging (MRI) showed a hyperintense lesion by T2-weighted image at the level of C4 without gadolinium enhancement (Figure 1). No lesions on dorsal MRI were found. T2-weighted fluid attenuated inversion recovery (FLAIR) MRI of the brain detected multiple hyperintense lesions (one nodular periventricular lesion and multiple punctiform juxtacortical lesions) (Figure 2). Visual evoked potentials were normal.
Laboratory evaluation findings, including routine chemistry, thyroid function, vitamin B12 level, vitamin E level, copper level, angiotensin-converting enzyme, erythrocyte sedimentation rate and C-reactive protein, were normal. Hepatitis B and C, human T-cell lymphotropic virus type 1 (HTLV-1), Lyme, syphilis and human immunodeficiency virus serologic test results were negative. Analysis of cerebrospinal fluid (CSF) found normal proteins content (0.28 g/L), normal glucose level (0.75 g/L), normal chlorine level (122 mmol/L), 2 white blood cells/mm3 and oligoclonal bands in immunoelectrophoresis. No oligoclonal bands were found in the serum. Neuromyelitis optica antibodies and myelin oligodendrocyte glycoprotein antibodies were negative. Antinuclear,
anti-centromere and anti-Sjögren’s syndrome-related antigen A (SSa) antibodies were positive. Lip biopsy did not show lymphocytic sialadenitis. The result of nailfold capillaroscopy was suggestive of systemic sclerosis.
The patient had a score of 10 according to The American College of Rheumatology/European League Against Rheumatism criteria for the classification of systemic sclerosis and a diagnosis of systemic sclerosis was established. The patient received five pulses of daily intravenous methyl-prednisolone (1 g) relayed by oral corticosteroids (prednisolone 20 mg/day) and monthly cyclophosphamide pulses with partial improvement after two pulses.
Discussion
Systemic sclerosis is a heterogeneous disease whose pathogenesis is characterized by three hallmarks: small vessel vasculopathy, production of autoantibodies, and fibroblast dysfunction leading to increased deposition of extracellular matrix.4 Our patient fulfilled the diagnostic criteria of systemic sclerosis according to the 2013 American College of Rheumatology and European League Against Rheumatism criteria.4 A minimum score of 9 is required to establish the diagnosis.4 The patient’s score of 10 was established by puffy fingers: 2, Raynaud’s phenomenon: 3, abnormal nailfold capillaries with capillary loss: 2, and systemic sclerosis-related autoantibodies anticentromere: 3.
This connective-tissue disease predominantly involves the skin, gastrointestinal tract, lungs, heart and kidneys. Unlike other collagen disorders, neurologic manifestations are considered uncommon, occurring in only 0.8–5.6% of patients and consisting mainly of muscle changes and peripheral neuropathy.1 CNS involvement is rare, with few case reports of cerebral arteritis.5 The most commonly reported symptoms are trigeminal neuralgia, migraine, tension-type headache and seizures.1
The association of neurologic abnormalities with systemic sclerosis has been considered incidental in some cases.6 In others, it has been related to iatrogenic causes, such as polymyositis secondary to steroids.1 In some cases, neurologic involvement was considered a complication of the disease itself, either by an indirect mechanism secondary to other organ involvement, like renal failure that can induce encephalopathy or neuropathy, gastrointestinal involvement that cause malabsorption and can induce myelopathy, peripheral neuropathy or seizures;7,8 or by a direct mechanism with primary pathologic processes in the nervous system.1 Hypotheses suggested in this latter case included vascular mechanism with endothelial changes and platelet aggregation that cause tissue ischaemia; dysimmune mechanisms considering the association of systemic sclerosis with other immune-mediated disease like thyroiditis, myasthenia gravis and insulin-dependent diabetes; and entrapment mechanisms caused by fibrotic processes in cases of peripheral neuropathy.1 Bertinotti et al. found, on proton magnetic resonance spectroscopy of patients with systemic sclerosis, that there was a reduction of N-acetyl-aspartate/creatine ratio and an increased choline/creatine ratio compared to controls.9 This is suggestive of neuroaxonal damage and CNS involvement in scleroderma.9 These spectroscopy data can be suggestive of inflammatory and demyelination mechanism.10,11
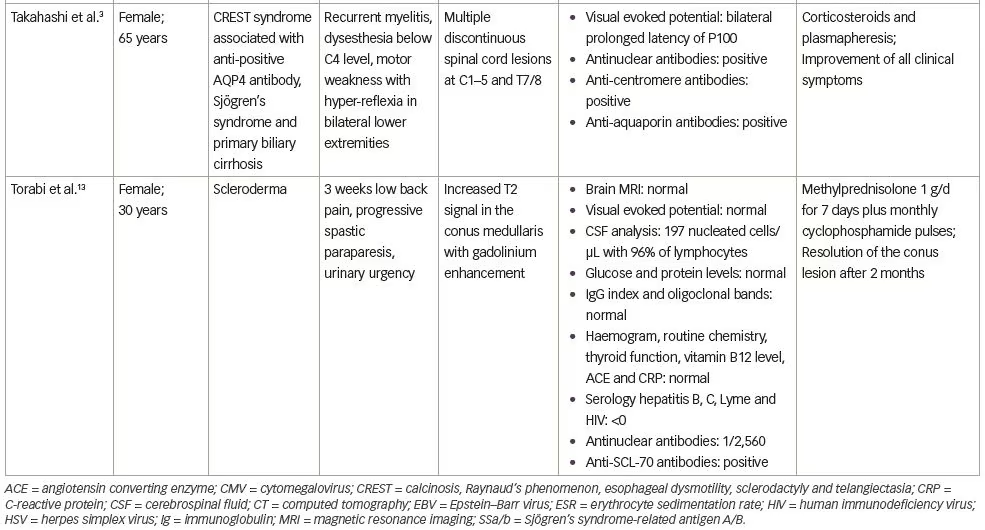
In this case, the ‘cause and effect’ relationship between myelopathy and systemic sclerosis was discussed. Spinal cord involvement was observed in 8% of cases in a series by Averbuch-Heller et al., which included 50 patients with scleroderma.1 In all patients, the diagnosis of scleroderma preceded the neurologic symptoms.1 Three patients had gastrointestinal tract involvement, but no metabolic deficit was identified.1 Observations of patients with scleroderma and myelopathy, found in the literature, are reported on Table 1.1–3,12,13
Spinal cord involvement may be manifested as cord compression secondary to mechanical osteolysis and facet arthropathy or massive calcific deposits.13 Considering the rarity of previous reports, and the absence of a compressive mechanism and gastrointestinal involvement with metabolic disorders that could explain the symptomatology in our patient, other possible inflammatory diseases were considered. Sjögren’s syndrome and MS in its primary progressive form were considered. Sjögren’s syndrome is associated with scleroderma in 10% of cases,1 and we found a positive result for anti-SSa antibodies; however, our patient did not fulfil the diagnostic criteria for Sjögren’s syndrome.14
For primary progressive MS, our patient fulfilled the 2017 updated McDonald diagnostic criteria.15 He had more than 1 year of disability progression; more than one T2 hyperintense lesions, characteristic of MS in periventricular and juxtacortical regions; and CSF-specific oligoclonal bands. Cases of scleroderma occurrence in patients already diagnosed with MS and treated by interferon beta are reported in the literature and the incrimination of this molecule in the development of the Scleroderma has been discussed.16,17 Association between MS and scleroderma is described in the literature in patients treated with interferon beta (IFN-β). In fact, IFN-β stimulates autoantibody production and induces autoimmune disorders.16 Only one case developed MS after scleroderma was reported in the literature.18
A rare allele in the CD45 gene has been found in relatively high frequency in both patients with MS and systemic sclerosis. This CD45 allele may enhance T-cell receptor signalling and may, thus, represent a genetic risk factor for both autoimmune diseases.16 Since there are no biomarkers for MS and the diagnosis cannot be retained, unless there are no better explanations for the clinical presentation, the question of whether this is systemic sclerosis revealed by a primary progressive MS-like condition or coexistence of systemic sclerosis and primary progressive MS in our case, remains open. In the absence of guidelines for the management of such patients and the possibility of cyclophosphamide use in the two diseases we chose to treat our patient with this molecule.
Due to therapeutic issues, it is important to consider systemic sclerosis in cases of MS-like conditions. In fact, the risk of a scleroderma renal crisis can be increased with steroid therapy; therefore, it is recommended to carefully monitor blood pressure and kidney function, and IFN should be also avoided.