Over the past two decades, monoclonal antibodies targeting the surface antigen CD20 have emerged as highly effective disease-modifying therapies (DMTs) for multiple sclerosis (MS).1 The major mechanism of action of these therapies is via B-cell depletion, as CD20 is expressed throughout much of the B-cell lineage, including pre-B-cells through memory B-cells. B-cells play multiple roles in the pathophysiology of MS, including pro-inflammatory and regulatory cytokine production, innate immunity, antigen trafficking and presentation, and antibody production, although the differential impact of these mechanisms remains uncertain.2
There are currently two anti-CD20 DMTs, ocrelizumab and ofatumumab, approved for the treatment of relapsing MS by the US Food and Drug Administration (FDA), with ocrelizumab also indicated for the treatment of primary progressive MS.3,4 These drugs also comprise the approved B-cell-directed DMTs, although their fellow anti-CD20 monoclonal antibody rituximab has been used extensively off-label.5 Ublituximab is a novel anti-CD20 monoclonal antibody that has recently completed the identical phase III ULTIMATE I and ULTIMATE II trials for relapsing MS.6 A literature search for English language articles and abstracts using the term ‘ublituximab’ was conducted on PubMed without date restrictions. The focus of this article is to provide a contextual overview of ublituximab’s pharmacology, review the available clinical data, and discuss the future considerations for its use as a DMT.
Pharmacological considerations
Anti-CD20 therapies lead to B-cell depletion, primarily in peripheral blood as opposed to lymphatic tissue, bone marrow or the central nervous system (CNS). In MS, this is thought to occur via two major mechanisms: antibody-dependent cell-mediated cytotoxicity (ADCC) via recruitment of natural killer (NK) cells and macrophages, and complement-dependent cytotoxicity (CDC).2 While current anti-CD20 monoclonal antibodies are among the most effective DMTs, they differ structurally (Figure 1), leading to variations in the relative contributions of each of these mechanisms to B-cell depletion, epitopes on CD20, immunogenicity and administration profiles (Table 1).7,8,9 Rituximab, ocrelizumab, ofatumumab and ublituximab are all type I antibodies, which localize CD20 to lipid rafts and have a corresponding high ability to mediate CDC when compared with type II antibodies.10 Ublituximab binds to an epitope on CD20 that is composed of two regions of the large extracellular loop, and is distinct from those regions bound by the other anti-CD20 DMTs (Table 1). An additional unique feature of ublituximab is that it is a glycoengineered monoclonal antibody (Figure 1). The fragment crystallizable (Fc) component of the antibody is responsible for binding complement component 1q (C1q) and Fcγ receptors. Glycans are heavily involved in structural fluctuations of the Fc conformation and resultant effector functions, notably the binding of FcγRIII that mediates ADCC.11 Ublituximab is manufactured with low fucose content in its Fc region, resulting in higher ADCC activity.12 In vitro assays using patient-derived chronic lymphocytic leukaemia cells demonstrated that ublituximab has ~100 times greater NK cell-mediated ADCC than rituximab.13 Given the elevated ADCC activity, it was proposed that ublituximab could be used at lower doses to treat MS, with fewer infusion reactions and shorter infusion times.
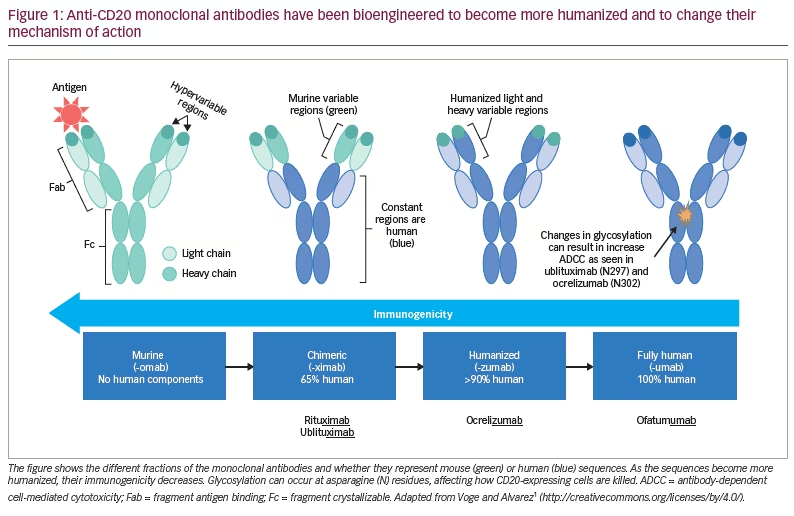
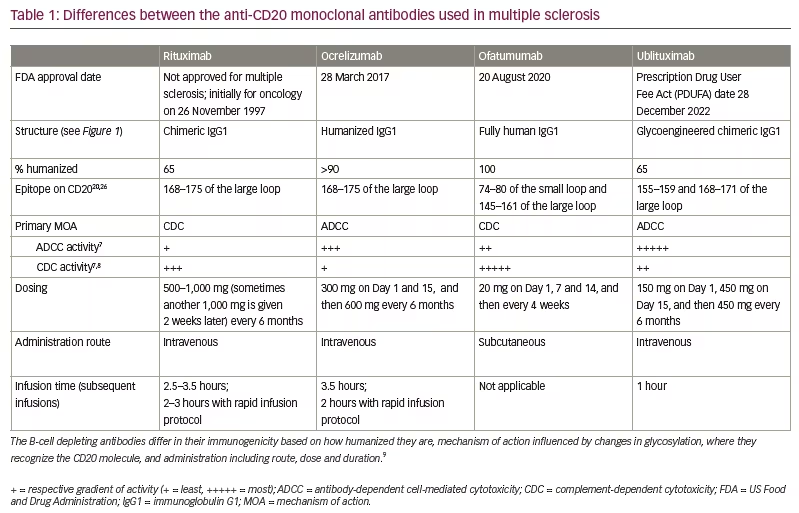
Like rituximab, ublituximab is a mouse–human chimeric antibody, while ocrelizumab is humanized and ofatumumab is fully human. When compared with fully human or humanized antibodies, this would be expected to lead to increased immunogenicity, with the generation of anti-drug antibodies. During the ULTIMATE studies, anti-ublituximab antibodies and ublituximab neutralizing antibodies were respectively seen at a rate of 17.8% and 2.4% at baseline or 86.5% and 6.4% at any point post-baseline.14 The presence of either type of antibody was not associated with higher relapse rates or infusion reactions. From a retrospective study of 339 patients, anti-drug antibodies may be found in approximately 33% of patients with relapsing MS treated with rituximab (the other chimeric anti-CD20 antibody), with their presence increasing with number of infusions and correlating with incomplete B-cell depletion, but not clearly with clinical measures of disease activity or infusion reactions.15 This frequency of anti-drug antibodies was similar to what was seen during the phase II HERMES trial in which 24% of patients had antibodies by 48 weeks of treatment.16 This uncertainty extends to the use of rituximab in the rheumatological literature, as well as to the use of ocrelizumab and ofatumumab, which are even less well studied. However, it is noted that there are no standardized assays for detecting antibodies, and differing time horizons and dosing across studies makes comparisons difficult.
In the ublituximab phase II trial, 100% of patients achieved the >95% endpoint reduction in CD19+ cells at 2 weeks, with a mean reduction of >99% and near complete depletion at 24 hours after the first infusion.17 This was maintained throughout the study and was long-lasting, with <25% reconstitution as patients entered the open-label ublituximab extension study (ClinicalTrials.gov identifier: NCT03381170) with a mean delay in treatment of 54.8 weeks.17–19 There was an accompanying reduction in pro-inflammatory CD4+ T helper 1 cells and increase in regulatory T-cells, as well as a reduction in pro-inflammatory memory T-cell populations. In the same trial, there was also an expansion of naïve T-cells.20 Taken together, these findings underscore the mechanistic benefits of anti-CD20-mediated B-cell depletion.
Clinical efficacy
Following initial development for the treatment of B-cell malignancies, ublituximab completed a promising phase II, dose-finding, placebo-controlled, randomized controlled trial (RCT) for relapsing MS in 2018, as partly described above. As described above, it achieved its primary end point of depleting >95% of B-cells at Week 4 (2 weeks after the second infusion).20 In this study, 93% of patients were free of either confirmed clinical relapses or 24-week confirmed disability progression (CDP).20 Radiologically, none of the patients receiving ublituximab had
contrast-enhancing lesions and 83% of the patients were free of new/enlarging T2 lesions. This resulted in 74% of the patients achieving no evidence of disease activity (NEDA). The annualized relapse rate (ARR) decreased by 95% from baseline to Week 48 and went from 1.45 to 0.07. Additionally, the mean T2-lesion volume decreased by 7.3% by Week 24 and by 10.6% by Week 48.20
Subsequently, the identical ULTIMATE I and ULTIMATE II phase III trials evaluating the use of ublituximab for the treatment of relapsing MS were undertaken.6 These studies were randomized, double blind, multicentre and active-comparator controlled with teriflunomide. Key inclusion criteria were typical for relapsing MS studies and included:
- age 18–55 years
- diagnosis of relapsing remitting MS with active disease (≥ 2 relapses in prior 2 years or one relapse in the prior year, and/or ≥ 1 gadolinium-enhancing lesion)
- an Expanded Disability Status Scale (EDSS) score of 0–5.5.
Prior treatment was allowed, with the exception of prior use of anti-CD20 monoclonal antibodies, alemtuzumab, natalizumab, teriflunomide, leflunomide or autologous haematopoietic stem cell transplantation. Patients in the ublituximab arm received an initial 150 mg infusion over 4 hours, a 450 mg infusion over 1 hour at Day 15, and then repeat infusions with an accompanying daily oral placebo at 24, 48 and 72 weeks. Patients in the teriflunomide arm received placebo infusions at the same time points while receiving daily 15 mg oral teriflunomide. Patients received antihistamine and corticosteroid premedication 30–60 minutes prior to infusions.6
Together, the ULTIMATE studies randomized 1,089 patients, 543 to ublituximab (271 in ULTIMATE I and 272 in ULTIMATE II) and 546 to teriflunomide (274 in ULTIMATE I and 272 in ULTIMATE II). Baseline demographics were well balanced between the studies and the treatment groups. The mean age was 35 and 37 years, respectively, for ULTIMATE I and II, with both groups having had 5 years since MS diagnosis and a mean EDSS score of 2.9. Overall, 64.2% of the participants were women and 98.0% were white, with 56.7% being treatment-naïve before starting the study.6
The primary endpoint was ARR at Week 96, for which ublituximab outperformed teriflunomide with an ARR of 0.08 versus 0.19 (rate ratio 0.41; 95% confidence interval [CI] 0.27 to 0.62; p<0.001) and 0.09 versus 0.18 (0.51; 95% CI 0.33 to 0.78; p=0.002) for reduction in clinical relapses of 59% and 49%, respectively, in ULTIMATE I and II.6 This effect extended to secondary radiographic measures, including the total number of contrast-enhancing lesions, with reductions in the ublituximab group of 97% and 96% (0.02 versus 0.49 [rate ratio 0.03; 95% CI 0.02 to 0.06; p<0.001] and 0.01 versus 0.25 [rate ratio 0.04; 95% CI 0.02 to 0.06; p<0.001]), respectively, in ULTIMATE I and II. Similarly, for the total number of new/enlarging T2 lesions, ublituximab resulted in decreases of 92% and 90% (0.00 versus 2.79 [rate ratio 0.08; 95% CI 0.06 to 0.10; p<0.001] and 0.28 versus 2.83 [rate ratio 0.10; 95% CI 0.07 to 0.14; p<0.001]). To evaluate disability, ULTIMATE I and II were combined and a non-statistically significant decrease was found at 12 weeks and 24 weeks, likely driven by the low number of events seen in both studies. Only 5.2% of patients receiving ublituximab and 5.9% of patients receiving teriflunomide had 12-week CDP (hazard ratio 0.84; 95% CI 0.50 to 1.41; p=0.51) while 3.3% and 4.8% had 24-week CDP, respectively (hazard ratio 0.66; 95% CI 0.36 to 1.21). Further analyses were not included in the hierarchical analyses, because this pooled analysis did not meet its endpoint. However, a prespecified pooled tertiary analysis found improvements in disability, with a 12-week confirmed disability improvement (CDI) of 12.0% in the ublituximab group versus 6.0% in the teriflunomide group (hazard ratio 2.16; 95% CI 1.41 to 3.31), and a 24-week CDI of 9.6% versus 5.1% (hazard ratio 2.03; 95% CI 1.27 to 3.25), respectively. Another secondary clinical measure included the percentage of patients achieving NEDA, which was defined as no clinical relapses, no magnetic resonance imaging (MRI) activity (new or enlarging T2 hyperintense lesions or T1 gadolinium-enhancing lesions),and no 12-week CDP. NEDA was achieved by 44.6% of participants on ublituximab and 12.4% on teriflunomide, from baseline to Week 96, a 3.6-fold improvement (p<0.0001).6 The rates of NEDA were improved to 82.1% in the ublituximab group versus 22.5% in the teriflunomide group when rebaselined from Week 24 to Week 96, maintaining a 3.6-fold improvement (p<0.0001); this was consistent across subgroup analyses between DMT-naïve groups versus prior DMT groups, and early versus late DMT experiences, early (within 3 years) MS diagnosis versus later diagnosis.21 This suggests that with higher efficacy therapies, NEDA is an achievable goal in most patients with MS.
Within the limitations of cross-trial comparisons, the identical OPERA I and OPERA II (ClinicalTrials.gov identifiers: NCT01247324 and NCT01412333) phase III RCTs evaluating ocrelizumab for relapsing MS reported an ARR of 0.156 and 0.155, respectively, in comparison to interferon β-1a 0.292 and 0.290 with decreases of 46% and 47%, respectively.4 The identical ASCLEPIOS I and II phase III RCTs evaluating ofatumumab for relapsing MS reported an ARR of 0.11 and 0.10 in comparison to teriflunomide 0.22 and 0.25, for a reduction of 50.5% and 58.5%, respectively.3 Although NEDA is more challenging to compare across trials given that it is a composite endpoint, it provides a meaningful, more comprehensive outcome for patients with MS. NEDA rates in OPERA I and II from weeks 24 to 96 were 41.9% with interferon β-1a versus 72.2% with ocrelizumab, a 72% improvement.22 The ASCLEPIOS studies with ofatumumab did not have an MRI at 24 weeks from which to rebaseline, but at Month 12, rates of NEDA with ofatumumab were 48.2% with teriflunomide and 87.7% with ofatumumab, an improvement of 82% percent.23 NEDA rates with ublituximab in the ULTIMATE trials were similar (described above in more detail), at 82.1% rebaselined from Week 24 to 96 and 88.2% from Week 48 to 96, although the improvements of 265% and 190% may suggest some degree of superiority compared with other anti-CD20s.21
Safety
A key aim of using ublituximab is to shorten infusion times while limiting infusion reactions. Common infusion reactions for anti-CD20 DMTs include fever, chills, rash, and hypotension, while anaphylaxis is rare. Overall, infusions are generally well tolerated, while ofatumumab (self-administered every 4 weeks via subcutaneous injection) has minimal reactions. Mechanistically, reactions occur due to rapid lysis of B-cells with resultant cytokine release, as suggested by more frequent infusion reactions when B-cells are present at the time of infusion, and from pre-medications themselves, such as sedation from antihistamines.24 In the ULTIMATE studies, 47.7% of ublituximab-treated patients reported an infusion reaction, with 43.3% of these occurring in the first infusion; most had no further infusion reactions compared with 12.2% of placebo infusions.6 Most reactions were mild, with 2.8% being grade 3 or higher and with two grade 4 reactions, one of which was anaphylaxis on the second dose. The most common reaction was fever in 9.5% of the ublituximab-treated patients, which may be related to the lack of acetaminophen as a premedication, although patients did receive steroids and antihistamines.6 During the first infusion of rituximab in the HERMES study, 78% of patients had infusion reactions, although there was no protocol for pretreatment with corticosteroids or antihistamines.16 In the OPERA studies, 34.3% of the ocrelizumab-treated patients had predominantly mild infusion reactions.4
There were no differences in rates of infections between ublituximab- and teriflunomide-treated patients in the ULTIMATE studies. Three deaths did occur in the ublituximab-treated participants due to pneumonia, encephalitis after measles, and salpingitis after an ectopic pregnancy.6 There are no available data on the clinical efficacy of vaccinations, including for SARS-CoV-2, in patients treated with ublituximab. It is likely that humoral response to standard vaccinations is impaired, as is seen with other anti-CD20 DMTs, and that this has clinical implications.25,26 The available data suggest that ublituximab treatment results in an enhanced naïve T-cell population that could respond to vaccinations and mediate some degree of protection.
Currently, there are limited data on other adverse effects of ublituximab infusions that are often seen with longer-term exposures, such as the rare reports of serum sickness seen with rituximab, and possibly ocrelizumab, based on a single case report.27,28 Adverse effects monitored closely with the anti-CD20 monoclonal antibody class, in addition to infusion reactions, are infections, hypogammaglobulinaemia and malignancy.2 Longer-term studies including real-world observations will be necessary for full accounting of these risks.
Neuromyelitis optica
While there are abundant data on the clinical effects of ublituximab in haematological malignancies (and several on-going studies), there has only been one other study focused on other CNS inflammatory diseases. A phase I open-label study for ublituximab (450 mg) as an add-on therapy to intravenous methylprednisolone, was undertaken for acute relapses of neuromyelitis optica spectrum disorder (NMOSD).29 There were no serious adverse events, but two patients relapsed at Days 58 and 81 after initial B-cell depletion. B-cell levels were <0.2% by Day 60, and 0.2% in one patient and 0.7% in the other patient at Day 90.29 How ublituximab may ultimately be used in NMOSD is not known, but there are theoretical suggestions based on the mechanisms of aquaporin-4–immunoglobulin G-mediated injury that increased ADCC activity would be beneficial.
Conclusions
Anti-CD20 monoclonal antibodies are well established as highly effective DMTs. With FDA approval likely to occur later in 2022, ublituximab will add another treatment option for relapsing MS. Goals of treatment should be identified prior to the selection of any MS DMT, and shared decision-making between the treating physician and the patient should focus on incorporating factors such as level of disease activity, risk tolerance, medical comorbidities, logistics (route and timing of DMT administration), and cost. While in the anti-CD20 class, ublituximab does have some differences from the other anti-CD20 DMTs. The quicker infusion compared with other DMTs, which is administered every 6 months, may appeal to patients and may help to reduce costs. However, rapid infusions over 2 hours with ocrelizumab from the ENSEMBLE PLUS study (ClinicalTrials.gov Identifier: NCT03085810) and the self-administered injections with ofatumumab, may create more competition in this therapeutic area where access and insurance coverage will become important.30 The relatively high ADCC activity suggests maintained efficacy at lower doses, which may improve efficacy in target tissues where penetration with antibodies can be limited, and is suggested by the slower repopulation of CD20+ cells. How this differentially impacts clinical efficacy and long-term safety is not yet known but will likely provide much discussion and debate in the years to come.