Acute disseminated encephalomyelitis (ADEM), first characterized in 1931,1 is a non-specific clinical syndrome of polyfocal central nervous system (CNS) inflammatory demyelination; it is characterized by encephalopathy and large, poorly demarcated cerebral white matter lesions.2,3 Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease (MOGAD), a specific neuronal antibody-mediated disease that can often present as ADEM, was discovered only in the last decade.4 Both occur more frequently in children than adults.3 With increasing awareness and testing for MOGAD, many children who historically lacked a clear aetiology for ADEM are being diagnosed with MOGAD.3,4 Although MOGAD can present with ADEM, MOGAD is a specific disease with its own diagnostic criteria, prognostic implications and therapeutic requirements.4 However, here at the Cleveland Clinic Neurological Institute, it is our experience that many clinicians unfamiliar with neuroinflammatory disorders of childhood continue to diagnose, document and manage confirmed MOGAD cases exclusively as ADEM, which can result in suboptimal care. The purpose of this review is, therefore, to clarify the differences between MOGAD and ADEM and provide practical updates based on real-world experience for the work-up and management of each.
ADEM can have numerous causes, with over 70% of cases preceded by infection.5–8 The estimated worldwide incidence of ADEM is 0.2–0.4 per 100,000 children annually, with up to 67% presenting in those younger than 10 years.1,2,6,8,9 In a large retrospective USA study of over 3,000 paediatric ADEM hospitalizations between 2006 and 2014, the overall incidence of ADEM cases and costs were found to be increasing across all ages, except in the Northeastern USA.9 Black, Hispanic and children from other ethnic minority groups showed higher incidences than Caucasian children. This discrepancy may be due to higher hospitalization rates from viral infections among these groups. The highest incidence of ADEM is recorded in winter and spring and the lowest in summer, likely reflecting seasonal variation in viral incidences.9
Interestingly, MOGAD has a similar incidence as ADEM, with approximately 0.3 cases per 100,000 children recorded annually worldwide.3,4 This number is likely to grow with increasing awareness of the disease. MOGAD presents most commonly as ADEM, optic neuritis or transverse myelitis.3–5 Cerebral cortical encephalitis and brainstem and cerebellar presentations are less frequent.3–5 In paediatric MOGAD, ADEM with or without optic nerve involvement is the most common initial presentation, particularly in children younger than 11.3–5 MOGAD can be monophasic, although 30–50% of children experience relapsing optic neuritis.3–5 No preferential racial or ethnic patterns have emerged to date.3–5 Proposed diagnostic criteria for paediatric MOGAD were recently published.4 Unlike ADEM, MOGAD does not require encephalopathy, although this can be present. Well over half of the children diagnosed with ADEM have MOG antibodies,10 although mode of testing, titre level and diagnostic criteria must be carefully weighed before MOGAD can be formally diagnosed. This is particularly important given the multitude of other causes of ADEM and the significant overlap between MOGAD, multiple sclerosis (MS), and neuromyelitis optica spectrum disorder (NMOSD).11,12
In our experience, a significant proportion of cases continue to be diagnosed as ADEM without specifying MOGAD as the underlying cause. In the aforementioned large ADEM cohort analysis, ADEM was a secondary diagnosis in the vast majority of cases (97.8%).9 When ADEM is not caused by MOGAD, it is more likely to be monophasic. On the other hand, MOGAD confers a significant risk of relapse.3,4 As discussed further below, failure to specify MOGAD as the cause of ADEM can delay targeted and timely treatment. Therefore, we have found it helpful to remove the ADEM diagnosis from a patient’s chart once MOGAD is confirmed to minimize confusion.
MOGAD overlaps with MS and NMOSD along a demyelinating disease spectrum. Features more characteristic of paediatric MOGAD than MS or NMOSD include bilateral, anterior, longitudinally extensive optic neuritis with optic nerve-sheath involvement and optic nerve–head oedema/haemorrhage; spinal cord conus medullaris lesions and ‘H-sign’ indicative of grey-matter involvement; pleocytosis of approximately 50 white blood cells/uL with mixed lymphocytes, neutrophils and monocytes; encephalopathy or encephalitis (including seizures); fever; headache; and positive meningeal signs.3,4,12-14
Unlike ADEM, MOGAD is a specific neuronal antibody-mediated disorder characterized by a significant elevation in MOG immunoglobulin (Ig) G1.3,4 The direct pathogenicity of this antibody remains unclear. MOG comprises the outermost layer of the myelin sheath, theoretically making it an easy target for antibody-mediated attacks. This could explain why MOGAD tends to be highly inflammatory, as demonstrated by the typical picture of a clinically severe attack; large, highly inflammatory, often oedematous magnetic resonance imaging (MRI) lesions; and a highly inflammatory cerebrospinal fluid (CSF) profile.3–5,13,14 This external surface-level accessibility might also explain why attacks are often so responsive to glucocorticoids, with quicker and more favourable functional recovery and resolution of lesions, compared with MS and NMOSD.3–5,13,14 It has been proposed that faster on-going myelination in young patients may confer greater antigenic heterogeneity, resulting in a wider spectrum of clinical presentation than that observed in adults.15
The Mayo Clinic Laboratories recently published validation studies on its cell-based serum MOG IgG1 assay.16 CSF sampling is presently less useful in MOGAD than in other conditions, such as NMOSD and N-methyl-D-aspartate-receptor encephalitis, where CSF titres can aid both in the diagnosis and treatment response.17,18 MOG IgG1 titres of at least 1:100 are consistent with MOGAD in the appropriate clinical context, as demonstrated by a positive predictive value of 82% overall.16 This value is probably even higher in children. The highest titre levels are observed when sampled close to the clinical event.19 Therefore, it is important to order the test as soon as MOGAD is suspected.
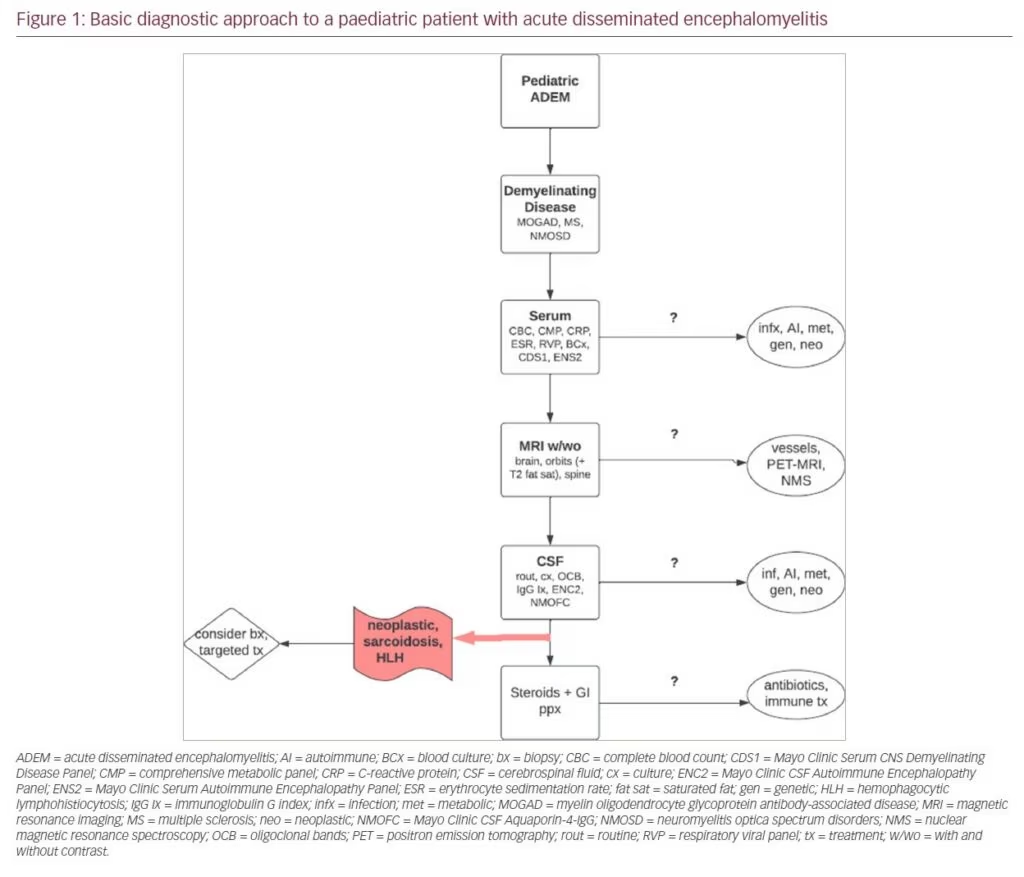
In our experience, it can take 1–2 weeks for validated antibody testing results to return. Therefore, it is critical to gather supportive evidence via other means to help facilitate appropriate and timely treatment. A typical workup of a paediatric patient with ADEM at our centre after initial emergency room triage and stabilization includes bloodwork followed by MRI and then lumbar puncture (Figure 1). Minimum serologic testing includes complete blood count, comprehensive metabolic panel, C-reactive protein, erythrocyte sedimentation rate, blood culture, respiratory viral panel, CNS Demyelinating Disease Evaluation (Mayo Test ID: CDS1) and Autoimmune Encephalopathy Evaluation (ENS2). Additional testing for infections and systemic autoimmune, metabolic, genetic, and neoplastic diseases should also be considered. MRI should include brain, orbits (with T2 fat-saturation sequencing), and whole spine, all with and without gadolinium. Dedicated vessel imaging, nuclear magnetic resonance spectroscopy and full-body positron emission tomography (PET)–MRI can also be considered if systemic autoimmune disease, vasculitis, metabolic disorders or neoplasm are suspected. Minimum CSF sampling includes routine analysis and cultures, oligoclonal bands, IgG index, Autoimmune Encephalopathy Evaluation (ENC2) and Neuromyelitis Optica (NMO)/Aquaporin-4-IgG Fluorescence-Activated Cell Sorting (FACS) Assay, Spinal Fluid (Mayo Clinic test ID: NMOFC). Additional infectious, metabolic and neoplastic evaluation may be required. If neoplasm, haemophagocytic lymphohistiocytosis (HLH), vasculitis or sarcoidosis is suspected based on clinical suspicion and the results of the workup described above, glucocorticoids may confound potential biopsy results, and alternative targeted therapies may be required.3,5–8,11
Recent evidence has suggested that prompt initiation of glucocorticoid treatment may improve paediatric MOGAD outcomes,20–22 although this requires further study. A recent Italian cohort analysis showed a 6.7-fold odds reduction in a relapsing course when glucocorticoids were administered less than 7 days from symptom onset.22 Regardless, it is our experience that early symptomatic recovery may be important, especially in children. Prolonged hospitalizations increase exposure to infections.23 In addition, slow recovery in a child can decrease a family’s confidence in their treatment team, potentially leading to a general reluctance towards further workup and therapies. It can also impact the likelihood that patients and families will engage in rehabilitation programmes and adhere to on-going follow-up, treatment and surveillance protocols.
When approaching a child presenting with potential ADEM, MOGAD should always be considered in the differential diagnosis. Excluding aetiologies for which glucocorticoid administration could be confounding shoud be prioritized. If the diagnosis of MOGAD is in question due to atypical clinical, radiologic or laboratory features, alternative aetiologies should be sought. Mimics and predisposing aetiologies include other demyelinating disorders, such as MS and NMOSD; infections; inherited immunodeficiencies; neoplasms; HLH; and metabolic, genetic and autoimmune diseases. If any underlying propensity to an exaggerated neuroinflammatory response is identified, it is possible that an infection caused a second hit, culminating in ADEM.6-9
Although glucocorticoids have not been shown to worsen many infections,24 excluding infection is nonetheless important in case antimicrobials are needed. Infection aetiologies linked to ADEM include measles, rubella, varicella–zoster virus, influenza, Epstein–Barr virus, herpes simplex virus, enterovirus, coxsackievirus, mycoplasma pneumonia, Borrelia burgdorferi, beta-haemolytic Streptococcus and SARS-CoV-2.25 If optic neuritis is present, Bartonella neuroretinitis should be considered.26 Consequently, it can be helpful to involve experts in infectious diseases to achieve targeted and accurate testing. Treatment of the infection concurrently with targeted immunotherapy for ADEM may be necessary. It is important to note that a subset of ADEM cases may yield false negative results in preliminary infectious workups;27 therefore, it is imperative to follow up on all final cultures, polymerase chain reaction tests and metagenomic next-generation sequencing.
Once diseases such as neoplasm, HLH, vasculitis and sarcoidosis have been reasonably excluded, glucocorticoids can typically be administered without delay. Acute therapy involves high-dose intravenous (IV) methylprednisolone at 30 mg/kg per dose daily (maximum daily dose 1,000 mg) for 3–5 days with gastrointestinal prophylaxis. Even if MOG IgG later returns negative, glucocorticoids can nonetheless be helpful, as many of the alternative causes of ADEM also respond favourably to steroid treatment.6,7
There are numerous other diseases that can predispose patients to ADEM. Systemic autoimmune diseases linked to ADEM include systemic lupus erythematosus, Sjögren’s syndrome, anti-neutrophil cytoplasmic antibody-associated vasculitis, scleroderma en coup de sabre with CNS involvement and Behcet’s disease.3 Inherited immunodeficiencies, such as combined variable immunodeficiency, and immune-pathway dysregulation, such as in complement factor I deficiency, can predispose patients to certain infections and lead to an exaggerated neuroinflammatory response.25,27,28 Potential mimics of ADEM include inborn errors of metabolism, such as lysosomal storage diseases and leukodystrophies.3,6,8 Genetic syndromes such as mitochondrial encephalopathy, lactic acidosis and stroke-like episodes and Aicardi–Goutières syndrome can result in a CNS vasculitis-type picture with inflammatory lesions.3,6,8,25,27
Some of these disorders can be reasonably excluded based on clinical history, physical examination and serologic testing. Others require a high index of suspicion and complex confirmatory metabolic and genetic analysis. A comprehensive investigation into family history, recurrent infections, rashes and joint symptoms, additional organ involvement, and neurocutaneous and neurovascular lesions can be invaluable in this regard. When evaluating for immune-pathway dysfunction by checking complement levels, lymphocyte subsets and other immunophenotyping, it is important to ensure that the patient has returned to baseline and that testing is performed prior to starting any long-term immunotherapy to avoid confounding.28 Initial metabolic screening with complete blood count with differential, liver and renal function tests, ammonia, blood gas, anion gap, glucose, lactate, and urine/blood ketones can be helpful.29 Rapid whole-genome sequencing is becoming more readily available due to lower costs and can help exclude a wide array of genetic disorders relatively quickly.30
Following recovery from an acute attack of MOGAD, we find it helpful to periodically re-evaluate patients for alternative aetiologies as appropriate, particularly before starting long-term immunotherapy. This may need to be done as an outpatient, as extensive laboratory testing can be challenging in the inpatient setting. In our experience, reasons for this include missed, incorrectly ordered or incorrectly performed tests (given these tests usually require larger quantities of blood and are more complex to order); the need for multiple blood draws in a young patient; and confusion over which tests to order due to the involvement of multiple subspecialists or the limited expertise in paediatric neuroinflammatory diseases.
Red flags of a more ominous underlying aetiology than MOGAD include clinical or radiological deterioration despite glucocorticoids or antibiotics, haemorrhagic lesions on susceptibility-weighted sequencing and progressive end-organ dysfunction.3,6,8,25,27,31 If any of these are present, it is important to evaluate for hypercoagulability and stroke risk factors, rare opportunistic infections, lymphoma and other neoplasms, paraneoplastic syndromes, HLH, Langerhans and non-Langerhans cell histiocytosis, and neurosarcoidosis. In such cases, an MRI of the chest, abdomen and pelvis or full-body PET-MRI can be helpful. Biopsies are sometimes required.3,6,8,25,27,31
One of the most challenging aspects of paediatric MOGAD is its relapse risk, which is estimated at 20–50%.3,4,31,32 Given the comparative greater risk of relapse and disability accrual with MS and the potential for severe irreversible neurological deficits with a first NMOSD attack, it is difficult to convey to families at initial diagnosis whether MOGAD will be lifelong and require long-term treatment. With a typical first clinically severe event, families are often eager to start acute treatment in the hopes of rapid symptomatic recovery. However, since neurological improvement can be more apparent in MOGAD in response to glucocorticoids compared with MS and NMOSD,3 it can be difficult for families to consider additional preventative treatment. Of note, disability scores with a first MOGAD attack have been shown to correlate with disability at follow-up.22,32 In contrast, the stigma of MS and its much higher risk of relapse are usually enough for families to do whatever is necessary to improve their child’s prognosis, including starting long-term treatment as soon as possible. This is one reason why starting a discussion about MOGAD at the first sign of a neuroinflammatory attack in a child can be very helpful. Families appreciate being given time to process and learn more about an unfamiliar diagnosis. It also allows them to start considering potentially complex long-term treatment options. After a second clinical attack, consensus generally favours starting long-term preventative therapy.4,22,33,34
Patients that relapse often do so in the first 6 months to 1 year following the initial attack and commonly after discontinuing glucocorticoids.22,32–34 Although data have been mixed regarding the optimal duration of prolonged oral glucocorticoid taper, recent reports favour at least 5 weeks.22,33,35–37 One would assume that continuing oral steroids for as long as possible would be indicated; however, their side effects in children can be severe.34 Therefore, if preventative neuroimmunotherapy is planned (usually with intravenous immunoglobulins [IVIg]37 or rituximab38), we generally start as early as possible. One strategy is to administer the first dose while the patient is still in the hospital following the acute attack and prior to discharge to allow for safety monitoring, as adverse events usually happen with the initial dose.39,40 Vaccination status also needs to be carefully considered, particularly in the context of rituximab therapy.41
For IVIg, one treatment option is to load 2 g/kg divided over either 2 or 5 days, followed by 1 g/kg monthly for at least 6 months (the maximum daily dose is usually about 100 g).37 Rituximab dosing can be of 750 mg/m2 (maximum daily dose 1,000 mg), with an initial load of 2 doses taken 2 weeks apart, then once every 6 months;38,41 however, no consensus protocol exists. The on-going COVID-19 pandemic combined with the start of the influenza and respiratory syncytial virus seasons may prompt a spike in neuroinflammatory disorders such as MOGAD in children during the winter months. In addition to the immunosuppression required to treat such patients, this means it will be especially critical for all children to stay up to date with age-appropriate vaccines to help minimize neurologic and infection-related morbidity and mortality.
IVIg seems to be the preferred option over rituximab for unvaccinated individuals diagnosed with MOGAD for whom long-term neuroimmunotherapy is planned and vaccination is suboptimal due to recent immunosuppression that can blunt vaccine response.42 It may confer added protection from infections since it consists of pooled IGs.43 On the other hand, B-cell-depleting therapies, including rituximab, increase the risk for patients of both contracting and experiencing severe complications from infections, including COVID-19.44 Previously, passive immunization with COVID-19 antibodies was available for added protection, but this is no longer recommended due to its failure to adequately protect against the newer common variants.45 In children, special consideration should also be given to vaccinations for conditions that are frequently overlooked, such as human papillomavirus.
If the decision is to remain on treatment, IVIg can be continued monthly. Alternatively, patients with MOGAD can be switched to rituximab. As with MS, we repeat the MRI 6 months after starting maintenance therapy; the new MRI scan serves as a new baseline scan against which future scans can be compared.14,41 We then proceed with the annual radiological surveillance, although it is unclear for how long in MOGAD, given its overall lower relapse risk and lesion accrual than MS and NMOSD.14,25 More long-term data are needed regarding the duration of maintenance immunotherapy. We, therefore, take a case-by-case approach by discussing with the family factors such as severity and frequency of attacks, overall disability, vaccination status, infectious risks, age, logistical treatment considerations, and socioeconomic burden.
In summary, paediatric ADEM is frequently caused by MOGAD. If atypical features or red flags exist, alternative aetiologies should be sought. However, prompt initiation of therapy is important, particularly in children. Until more long-term studies are undertaken to explore this important disease entity further, management decisions should be rooted in consensus expert recommendations, combined with an objective, stepwise, thoughtful approach individualized for each patient with close involvement of the patient’s family. This will help improve care and optimize outcomes while awaiting the results of further investigation.