The signal interpreted as pain can be altered at many points along the pain pathway, providing a multitude of strategic targets for rational therapeutic intervention. However, despite the huge increase in our understanding of the cellular and molecular mechanisms that initiate and maintain maladaptive pain processing, chronic pain remains a huge unmet clinical need. The pharmacopeia of available drugs is effective in some chronic pain patients, but not all of the time, due to disease progression, low efficacy, and, more often, intolerable side effects. Thus, efforts need to continue to boost the pharmaceutical profile of available agents by improving efficacy, tolerability, and pharmacokinetics, in addition to developing new chemical entities to manage pain refractory to current drugs. This article will focus on some of the molecular mechanisms and targets that are currently attracting interest in chronic pain research.
Excitatory Mechanisms—Blocking Aberrant Activity
Voltage-gated Sodium Channel Blockers
Sodium channel (NaV) blockers such as local anesthetics, carbamazapine, etc. have been the mainstay of pain medicine for decades, producing anesthesia and analgesia by preventing action potential propagation; however, blocking conduction is not their only role. Three of the nine isoforms of the human sodium channel family, NaV 1.7, 1.8, and 1.9, are expressed selectively in damage-sensing peripheral neurons.1,2 Animal studies have demonstrated a role for these channels in mechanosensation. Accordingly, mechanical hypersensitive behaviors akin to allodynia (painful experience to innocuous stimulation, e.g. touch) and hyperalgesia (heightened pain experience to painful stimuli) seen in chronic pain models are attenuated in tissueselective knock-out mice lacking these selective pain-related channels.3
Aberrant activity in damage-sensing peripheral fibers following inflammation arises from the processes of peripheral sensitization, whereas clustering of these channels at the site of injury and neuroma after nerve injury is thought to be a major drive for the induction of disordered central processing.3 The newly dense distribution along the sensory neurone supports spontaneous firing of action potentials along the neurone in the absence of peripheral stimuli. Additionally, a concomitant upregulation of the embryonic NaV 1.3 occurs, whose biophysical properties and re-expression in adult neurones after nerve injury have been associated with increased neuronal excitability and neuropathic pain.4 However, a recent study has refuted its role in nerve-injury or chronic inflammatory pain,5 although its role in central neuropathic pain could be considered on the basis of its upregulation in thalamic nuclei.6 Pre-clinical data point to a major role for NaV 1.7 in acute and inflammatory pain states.7 Differential mutations in the encoding gene, SCN9A, have been linked to the intense burning pain in erythermelalgia patients, a congenital absence of pain perception, and reduced lidocaine effectiveness in some pain patients.8,9 Understanding the genetic basis for altered nociceptive profiles in chronic pain patients could lead to selective therapy on an individual basis. A critical consideration for utilising the therapeutic benefit of NaV blockers is a clear separation between cardiovascular and sensory neuronal effects without numbness, which requires subtype selectivity. A potent and selective isoform-specific small-molecule (NaV 1.8) blocker (A- 803467) that reduced inflammatory and neuropathic pain behaviors in rats is in development, and represents a potentially exciting new treatment for allodynia and hyperalgesia in chronic pain patients.10,11 Other promising new treatments include lacosamide and ralfinamide. Antinociceptive efficacy for the anticonvulsant lacosamide was shown in models of chronic inflammation and neuropathic pain, and has translated into promising results in phase II and III clinical trials for painful diabetic neuropathy; the drug produced marked reductions in pain scores compared with placebo and was generally well tolerated, and low drug–drug interactions were reported.12,13 Unlike other anticonvulsants, the drug has a novel dual mode of action, whereby it enhances slow inactivation of voltage-gated sodium channels and interacts with collapsin-response mediator protein-2.14 This possibly sets the drug apart from other sodium-channel-blocking drugs such as lamotrigne and topimarate, which have proved disappointing in treating painful diabetic neuropathy.15,16 Ralfinamide is proposed to inhibit NaV 1.3, 1.7, and 1.8 and CaV 2.2 currents in rat dorsal root ganglia (DRGs) to suppress evoked and spontaneous pain in chronic inflammatory and neuropathic pain models, and positive phase II trials in neuropathic pain patients have been reported.17 Understanding how alterations in NaV channels contribute to the pathogenesis of chronic pain conditions should enable selective blockers to be produced that have therapeutic efficacy at doses well below those that impair nerve conduction or cardiovascular function, among other side effects.
Calcium Channels
Voltage-dependent calcium channels (VDCCs) are important for both transmitter release from peripheral afferents and neuronal excitability. Ten subtypes have been cloned, many of which are critical in pain regulation. The N-type (CaV 2.2) is of particular interest since its activity is enhanced in chronic pain states.18 Genetic deletion results in an attenuation of inflammatory and neuropathic pain behaviors, and the selective blocker ziconotide has proved successful in both animals and human studies.19,20
To date the most clinically successful modulators of Ca2+ channel activity are agents targeting the α2δ-1 and 2 subunit of high-voltage Ca2+ channels, with antinociceptive efficacy seen in cancer/ chemotherapy, HIV, and diabetic neuropathies and osteoarthritis.21–24 Furthermore, a marked upregulation of α2δ-subunits has been correlated with the onset and development of allodynia and hyperalgesia and the effectiveness of α2δ ligands. Gabapentin and its newer analog pregabalin are first-line treatments for neuropathic pain. Additional α2δ-ligands with improved bioavailability and efficacy are in development.
Hyperpolarization-activated Cyclic Nucleotide-gated Channels
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, which generate spontaneous rhythmic activity in the cardiovascular and nervous systems, play an important role in modulating neuronal excitability and plasticity in chronic pain states.25 Nerve injury results in increased HCN1 expression in DRGs and spontaneous activity in the damaged nerve. Studies using the selective antagonist ZD7288 decreased ectopic activity in DRGs and reversed spontaneous pain and allodynia induced by mild thermal injury and nerve injury. Thus, drugs targeting HCN currents could prove beneficial for chronic pain treatment.26,27 Transient Receptor Potential Channels
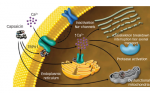
The first cloned nociceptor was identified as the transient receptor potential (TRP) V1 channel.28 Expressed at sensory nerve terminals, this ligand-gated nociceptor can integrate various painful stimuli, e.g. capsaicin, low pH, and noxious heat, and may underlie neuronal hypersensitivity due to modulation by inflammatory and nerve-damageassociated mediators, e.g. prostaglandins, bradykinin, and nerve growth factor (NGF).29 Paradoxically, antinociception is achieved with agonists via receptor desensitization, e.g. topical high-dose capsaicin is effective in neuropathic patients, and also with antagonists. In the latter case, the efficacy of antagonists relies on the promiscuous nature of the receptor since blocking heat rather than mechanical responses is unlikely to be clinically important.30,31 Caution is needed due to the potential for altered body temperature.
An intriguing recent development utilizes TRPV1 to enable the delivery of a lidocaine derivative to specifically block damage-sensing neurons without blocking all nerve cells, thus avoiding paralysis and numbness.32 Here, activation of TRPV1 is used to induce opening of Na channels, thus enabling better access for the lidocaine. This strategy would most likely be useful peri- and post-operatively for surgical pain, but similar therapies could eventually be developed for chronic pain treatment. Other TRP channels of interest include TRPV3, TRPV4, and TRPA1, which are upregulated in neuropathy and sensitized by inflammatory mediators.33,34 TRPM8, the receptor for cooling found in sensory nerves, transduces cold hypersensitivity following inflammation and nerve injury; paradoxically, this channel also produces profound analgesia in chronic pain states, including neuropathic pain.35
N-methyl-D-aspartate Receptor Antagonists
The N-methyl-d-aspartate (NMDA) receptor for glutamate is a complex protein that requires binding of two co-agonists as well as a long-lasting membrane depolarization to enable receptor activation. Other regulatory sites include polyamine and phencyclidine (PCP) binding sites. NMDA-R activation underlies wind-up, a measure of neuronal hyperexcitability, and the increased responsiveness of spinal neurones is probably the basis for central sensitization, which likely underlies the increased pain and area of pain that are commonly observed after injury.36
Chronic inflammatory and neuropathic pain states involve NMDA-R activation, and antagonists at multiple sites are effective in a number of animal models of pain and in pain patients, but the small therapeutic window (psychotomimetic effects at analgesic doses) means that antagonists have not been as successful as pre-clinical studies first indicated.37 However, antagonists such as ketamine act synergistically with opiates, either by enhancing their antinociceptive effect or by blocking analgesic tolerance;38 therefore, this route of clinical intervention still holds promise for chronic pain management.
NR2B subunit-containing NMDA-Rs are selectively expressed in sensory pathways, and the antinociceptive efficacy of NR2B blockers has been shown in a range of pre-clinical chronic pain models without the psychotomimetic effects that have plagued other non-selective NMDA blockers.39 Additionally, synergistic effects with an NR2B antagonist and 3-methyl-gabapentin have been reported in neuropathic mice.40 Thus, these findings have renewed research efforts in the therapeutic utility of NMDA receptor antagonists.
Descending Facilitation
A critical role for descending facilitation, originating from brainstem areas, in the development and maintenance of chronic pain states has been demonstrated.41–45 One such pro-nociceptive pathway originates from spinal neurokinin1-expressing projection neurons, relaying through brain nuclei, which in turn modulate descending monoaminergic pathways from the brainstem back to the cord, forming complex spino–bulbo–spinal loops.46 Activation of the loop engages the rostroventral medial medulla to control spinal excitability through the activation of a descending serotonergic drive, which is bi-directional. Thus, in addition to inhibition, the brain can amplify spinal pain processes via pro-nociceptive spinal 5-HT3 receptor activation.46,47 There is evidence for an overdrive or inappropriate activation of descending serotonergic (5-HT3) facilitation in models of nerve and spinal cord injury and cancer-induced bone pain.45,48–50 Interestingly, an intravenous injection of the selective 5-HT3 receptor antagonist ondansetron reduced pain scores in neuropathic patients compared with placebo.51 Similar evidence exists for fibromyalgia patients, in whom the 5- HT3 antagonist tropisetron produced significant reduction in pain scores.52,53 Enhancing Inhibitory Mechanisms
Potassium Channel Blockers
A feature of chronic pain is neuronal hyperexcitability. Potassium (K) channels set resting membrane potentials and re-polarize action potentials, thus limiting neuronal excitability. The anticonvulsant drug retigabine, currently in phase III clinical trials for epilepsy, is effective in pre-clinical pain models.54–56 The drug acts as a ‘brake’ on repetitive firing that leads to neuronal hyperexcitability by opening KV 7 channels. Accordingly, retigabine produced a greater inhibitory effect on neuronal hyperexcitability and attenuated behavioural hypersensitivity in neuropathic rats; therefore, there is interest in developing this compound for the clinical treatment of neuropathic pain.
Other K channels of interest to pain processing include the KCNK potassium channel family. The active ingredient in szechuan peppers was recently shown to excite sensory neurons by inhibiting KCNK3, 9, and 18 channels.57 Additionally, reduced thermal and mechanical inflammatory hyperalgesia was observed in KCNK2 (or TREK-1) mutant mice.58 These KCNK channels co-localize with TRPV1 DRG (peripheral nociceptors) and are sensitive to volatile anesthetics.59 Whether exploiting K-channel activity will yield effective tolerable drugs for chronic pain treatment remains to be seen, but current findings suggest that these channels merit further investigation.
Monoamines
Antidepressant drugs, which include tricyclics and selective serotonin and dual-action serotonin and noradrenaline re-uptake blockers (SSRI/SNRIs), have proved beneficial in the treatment of chronic pain, acting primarily to enhance plasma concentrations of serotonin and noradrenaline.60 Current findings point to a greater effectiveness of drugs that either enhance both systems and/or have dominance for enhancing activity in noradrenergic activity, e.g. duloxetine (SNRI) rather than the SSRIs, which may in part be due to the bi-directional effects of the serotonergic system. The actions of these drugs are independent of their antidepressant actions and more likely exert their pain-relieving effects via enhancement of descending inhibition.61
Targeting Non-neuronal Cells—Neuroimmune Modulation
Mounting evidence implicates the critical role of immune and glial cell activation and hypertrophy in the development of chronic pain. These cells release a number of pro-nociceptive mediators such as cytokines, neurotrophins, nitric oxide, etc. that can amplify the pain signal and spread hyperexcitability by altering neuronal activity and immune cell interactions.62 Thus, glial inhibitors have proved beneficial in pre-clinical pain models; for instance, a marked upregulation of spinal purinergic P2X4-R, expressed by microglia, occurs after nerve injury, and P2X4 antagonists reverse the ensuing allodynia/hyperalgesia in these animals.63 The antibiotic minocycline also produces neuroprotective and anti-inflammatory effects via its inhibitory actions on microglia, and the astrocyte inhibitor propentofylline has been shown to attenuate mechanical allodynia.64,65
Chemokines are expressed and upregulated in many pain states. In particular, chemokine monocyte chemoattractant protein-1 (CCL2) triggers spinal microglia activation in neuropathic conditions and holds promise as a selective target due to its preferential action at the chemokine receptor CCR2.66 CCL2 neutralization partially reverses mechanical hyperalgesia and CCR2-/- mice have no mechanical hyperalgesia after nerve injury.66,68
Not all immune-cell-evoked mediators are pro-nociceptive. In particular, interleukin (IL)-10 blocks the production, release, and actions of tumor necrosis factor, IL-1, and IL-6 to produce a marked reduction in allodynia and hyperalgesia in chronic pain models.65,66 However, since IL-10 cannot cross the blood–brain barrier and has a short half-life, systemic administration has not proved useful. A gene transfer delivery system using viral vectors improved matters, but elicits only a transient block of allodynia and hyperalgesia lasting about two weeks. More recent developments using synthetic polymers to deliver plasmid DNA encoding IL-10 resulted in improved potency and longlasting (three-month) pain relief.71,72
These and many other studies highlight the importance of controlling aberrant glial activity while enhancing the beneficial effects of some cytokines in chronic pain states. Disease Modification—Reversing Pathology
The available therapeutic agents block part(s) of the pain pathway, thus alleviating pain symptoms but not necessarily reversing the pathological adaptations that maintain pain. Disease-modifying agents that could reverse the pathology that begets chronic pain rather than just symptom management would provide a better strategy. A molecule with the potential to do just that is the neurotrophic factor artemin, a member of the glial-derived neurotrophic factor (GDNF) family that promotes neuronal survival. Artemin reversed allodynia and hyperalgesia in neuropathic rats, together with partial to complete normalization of many morphological and neurochemical features of nerve injury, which was effective for as long as artemin was administered.73 Interestingly, artemin returns normal sensory function, including nociceptive function, after sensory loss in a model of brachial plexus injury, with positive effects persisting for six months after treatment cessation.74 Thus, these findings represent a big step forward towards achieving both pain relief and reversal of the pathophysoiological mechanisms maintaining chronic neuropathic pain.
There is an abundance of evidence linking NGF to pain generation and central sensitization evidenced in several animal and human pain studies.75 Expressed in primary sensory neurones, NGF is upregulated in models of chronic pain and in patients with arthritis, pancreatitis, and inflamed bladders, and in turn upregulates many painrelated transmitters and receptors—e.g. substance P, TRPV1, sodium channels, etc.—to enhance pain.75–79 NGF antagonism prevents the onset of allodynia and hyperalgesia in many animal pain models, and, perhaps more importantly, provides relief in established models of inflammatory and neuropathic pain.80 Recently, a humanized monoclonal antibody against NGF (Tanezumab, Pfizer) has proved successful in phase II trials for the treatment of pain related to knee osteoarthritis.
Conclusion
The pathophysiology of chronic pain involves myriad complex nociceptive mechanisms and pathways and therefore has remained difficult to treat, with current therapies lacking sufficient efficacy and/or having intolerable side effects. This review provides a snapshot of some of the key molecular targets, i.e. ion channels, TRPV1, monoamines, glia, and neurotrophic factors, that hold the most promise for future chronic pain therapy. Continuing efforts to discover the mechanisms involved in chronic pain will allow the rational development of selective agents, which by virtue of their precise nature of activity should in time generate better analgesics.