
Postpartum depression (PPD) is an important mental health problem that is associated with maternal distress and poor maternal–fetal bonding. The Diagnostic and Statistical Manual of Mental Disorders (DSM-5) now classifies PPD as major depressive disorder (MDD), with peripartum onset.1 The distinction ‘with peripartum onset’ is new in DSM-5 and is important, as 50% of ‘postpartum’ major depressive episodes actually begin before delivery. Prevalence estimates of major depressive episodes with peripartum onset vary between 3% and 6%1 and are as high as 10–20%.2 The cause of PPD has yet to be fully elucidated. Maternal neurotransmitter abnormalities, neuroendocrine changes, neuroinflammation, rapid drop in estrogen levels after delivery, hypothalamic–pituitary–adrenal axis dysfunction, thyroid dysfunction and genetic predisposition have all been proposed as contributing factors.2,3
The most commonly prescribed antidepressants for PPD are selective serotonin reuptake inhibitors (SSRIs) and serotonin and norepinephrine reuptake inhibitors. While conventional antidepressants are generally safe and well tolerated in the postpartum period, they take up to 4–6 weeks to work. Electroconvulsive therapy (ECT) is a well-studied, safe, faster alternative to conventional antidepressants. One study found that 34% of patients remitted by Week 2 of ECT and 60% of patients remitted by Week 3.4 Unfortunately, ECT requires an ECT treatment suite and an anaesthesiologist, which are not available in all areas of the country. ECT initiation also typically requires three treatments per week for several weeks, and sometimes also an inpatient hospitalization, which is often not a desirable option for new mothers.
The limitations of existing antidepressant treatments have led to a search for medications that act faster, especially in the postpartum period when crucial maternal–infant bonding is occurring. There has been recent interest in both ketamine and esketamine as novel approaches to treating MDD. While the rapid effects of these drugs are promising, the effects are short lived without repeated administration; after the acute response, ketamine is usually administered every 1–3 weeks to maintain a response.5 The need for repeated administration raises concern for long-term side effects of these medications, as well as abuse potential. It is also worth noting that the mechanism of action for the antidepressant effects of ketamine and esketamine is not fully understood. It was previously thought that the antidepressant effects of ketamine were mediated through N-methyl-D-aspartate receptor antagonism and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid modulation; however, a recent study found that the antidepressant effects of ketamine are blocked when naltrexone, an opioid antagonist, is given before ketamine administration.6 This suggests that the antidepressant effects of ketamine are dependent on opioid receptor activation and that ketamine is not fully addressing the underlying neurobiological cause of depression.
Recently, the neuroactive steroid (NAS) allopregnanolone has been implicated in depressive disorders in both women and men. Sex differences in the prevalence of affective disorders suggest that sex steroids play an important role in the development of mood disorders.7 Patients with depression have lower cerebrospinal fluid, serum and plasma allopregnanolone levels than their counterparts who are not depressed.7 Furthermore, MDD has been shown to be associated with lower allopregnanolone levels whereas antidepressant treatment increases allopregnanolone levels.2
Allopregnanolone levels change during the peripartum period.8 Levels increase during pregnancy, leading to downregulation of gamma aminobutyric acid A (GABA-A) receptors. Following delivery, estrogen and allopregnanolone levels drop quickly and reach pre-pregnancy levels within 3 days. GABA-A receptors are slow to adapt to these lower levels and sensitivity to these changes have been implicated in the aetiology of PPD.
GABA has also been implicated in MDD without peripartum onset. Similarly to allopregnanolone, GABA levels are also reduced in the cerebrospinal fluid and plasma in people with depression.9 One study demonstrated that unmedicated patients with depression have reduced GABA in the occipital cortex and another study found a reduction in GABA concentrations in the prefrontal cortex.10,11 Additionally, in people with depression who died by suicide, concentration of mRNA that encodes for the α4 and δ extrasynaptic GABA-A receptors is reduced.
Pharmacologically, allopregnanolone is a progesterone metabolite and NAS that acts as a GABA-A receptor positive allosteric modulator (PAM). GABA is an inhibitory neurotransmitter in the brain that is present on 30–50% of neurons.12 GABA and allopregnanolone produce both synaptic (phasic) inhibition, the site for benzodiazepine binding, and extrasynaptic (tonic) inhibition at the GABA-A receptor.13 GABA-A receptors are especially sensitive to tonic inhibition by allopregnanolone and the tonic effect is thought to mediate the long-lasting impact of allopregnanolone and its analogues. This article will review available data on the two NAS drugs with significant research – brexanolone and zuranolone.
Search strategy
A systematic literature search was conducted in PubMed, Ovid MEDLINE and Cochrane databases for any publications containing the following keywords: neuroactive steroids, NAS, gamma-aminobutyric acid, GABA-A, positive allosteric modulator, PAM, postpartum depression, allopregnanolone, brexanolone, zuranolone, ganaxolone and PRAX-114. ClinicalTrials.gov was accessed for posted NAS GABA-A receptor PAM studies and results. Peer-reviewed presentations and posters at major national and international psychiatric meetings were also identified.
Brexanolone
Proof-of-concept study
The utility of brexanolone was first demonstrated in an open-label, proof-of-concept study performed at the University of North Carolina Perinatal Psychiatry Inpatient Unit.14 Four women who met criteria for a severe episode of MDD (Hamilton Depression Rating Scale-17 item [HAMD-17] ≥20) that started between the beginning of their third trimester of pregnancy and 12 weeks after delivery were enrolled. All patients were administered brexanolone at doses that mimicked physiological levels of allopregnanolone. Brexanolone was titrated over a 12-hour period to a maintenance dose of 86 µg/kg/h. The maintenance dose was administered at this rate for 36 hours, and then tapered over a period of 12 hours (total of 60 hours). Adverse events (AEs) were reported by all four patients; all were mild to moderate in severity. Sedation was the most common AE reported. Mean HAMD-17, Edinburgh Postnatal Depression Scale (EPDS), Patient Health Questionnaire (PHQ-9) and Generalized Anxiety Disorder-7 (GAD-7) scores were reduced from pre-treatment baseline at all timepoints assessed in the study, from 12 hours to 84 hours.
Study 202A (NTC02614547)
Following the positive proof-of-concept study, this phase II double-blind trial randomized 21 women aged 18–45 years with PPD to receive either brexanolone 90 µg/kg/h or placebo.15 Ten patients were randomized to the brexanolone group and 11 were randomized to placebo. All patients met criteria for a major depressive episode (HAMD-17 ≥26) that started between the beginning of their third trimester of pregnancy and 4 weeks following delivery. All women were less than 6 months postpartum and were allowed to be on stable antidepressant therapy provided the dose was not changed during the study. Participants had to agree not to start any new medications during the study, including anxiolytics as needed. Women with active psychosis, history of seizures, bipolar disorder, schizophrenia, schizoaffective disorder and history of substance abuse within 12 months of study commencement were excluded. Women with a history of attempted suicide during their first episode of PPD were also excluded.
Baseline demographics were similar across both groups, with 30% of women in the brexanolone group and 27% of women in the placebo group taking antidepressants. All patients received an infusion of the assigned study drug over 60 hours while under inpatient care according to the following schedule: 30 µg/kg/h (0–4 hours), 60 µg/kg/h (4–24 hours), 90 µg/kg/h (24–52 hours), 60 µg/kg/h (52–56 hours) and 30 µg/kg/h (56–60 hours). HAMD-17 and Montgomery–Asberg Depression Rating Scale (MADRS) scores were measured at 0 hours, 12 hours, 24 hours, 36 hours, 48 hours, 60 hours, 72 hours, 7 days and 30 days post-infusion. The primary outcome in this study was the reduction in HAMD-17 from baseline at 60 hours post-infusion. Secondary outcome measures were the proportion of patients achieving remission (HAMD-17 total score ≤7) and response (≥50% reduction in HAMD-17 total score). The primary outcome was met, with a mean reduction in HAMD-17 score of 21 points in the brexanolone group compared with 8.8 points in the placebo group (p=0.0075).
Several of the secondary outcome measures were also met (Table 1).
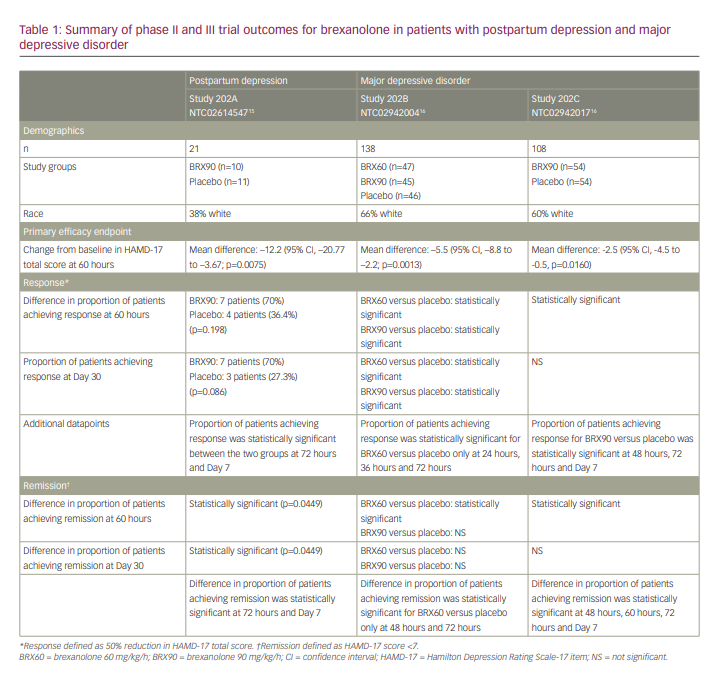
Beginning at 24 hours, more patients in the brexanolone group experienced remission of symptoms compared with the placebo group. This difference was statistically significant at 24 hours and was maintained at 30-day follow-up. Response in the brexanolone group was numerically greater than the placebo group (70% versus 36.4%) at 60 hours; however, this difference was not statistically significant at this timepoint. The difference in response in the brexanolone versus placebo groups did become statistically significant at 72 hours (80% versus 27%; p=0.0374) and this difference was maintained at Day 7 (80% versus 20%; p=0.0335) and Day 30 (70% versus 27.3%; p=0.086) follow-up.
Brexanolone was well tolerated, with 4/10 patients in the brexanolone group and 8/11 patients in the placebo group reporting AEs. The most frequently reported AEs in the brexanolone group were dizziness and somnolence. Other AEs reported in the brexanolone group included rash, postural dizziness, dry mouth, hot flush, pyrexia, sinus tachycardia and vertigo. Sinus tachycardia and somnolence were classified as moderate treatment-emergent AEs in the brexanolone group. There were no serious AEs or AEs that led to drug discontinuation.
Studies 202B (NTC02942004) and 202C (NTC02942017)
Studies 202B and 202C are both US-based phase III multicentre trials.16 Study 202B enrolled women with a severe episode of MDD (HAMD-17 >26) while Study 202C enrolled women with a moderate episode of MDD (HAMD-17 20–25). Inclusion and exclusion criteria were the same for both studies. All patients were aged between 18 and 45 years and had onset of the depressive episode during the peripartum period. Exclusion criteria included renal failure requiring dialysis, fulminant hepatic failure, anaemia, known allergy to allopregnanolone or progesterone, active psychosis, history of schizophrenia, bipolar disorder or schizoaffective disorder, history of suicide attempt during current depressive episode and history of alcohol or drug abuse in the previous 12 months. Women were also excluded if they had undergone ECT treatment within 14 days of screening or planned to start ECT within 7 days after their infusion.
Baseline demographics were similar across all groups in both studies. Notably, 22% of 246 patients across both studies were taking antidepressants at baseline. The primary efficacy outcome in both studies was the change from baseline in mean HAMD-17 total score at 60 hours post-infusion. All patients were followed up to Day 30. Infusions were given over 60 hours according to the following schedule for the BRX90 group: 30 µg/kg/h (0–4 hours), 60 µg/kg/h (4–24 hours), 90 µg/kg/h (24–52 hours), 60 µg/kg/h (52–56 hours), 30 µg/kg/h (56–60 hours). Patients in the BRX60 group received their infusion according to the above schedule, except they received their infusion at a rate of 60 µg/kg/h from 24–52 hours.
A total of 138 women were enrolled in Study 202B and were randomized 1:1:1 to receive placebo (n=46), brexanolone 60 µg/kg/h (n=47) or brexanolone 90 µg/kg/h (n=45). The reduction in HAMD-17 total score was 19.5 points in the BRX60 group, 17.7 points in the BRX90 group and 14 points in the placebo group at 60 hours post-infusion initiation (primary endpoint). The difference in the change in HAMD-17 score reached statistical significance in both the BRX60 (p=0.013) and BRX90 (p=0.0252) groups at the primary endpoint. Remission data in this study showed that more patients in both the BRX60 and BRX90 groups achieved remission at 24 hours, 36 hours, 48 hours, 60 hours, 72 hours and Day 30 than those receiving placebo. This difference achieved statistical significance in the BRX60 group at 48 hours, 60 hours and 72 hours; however, the remission rates were not statistically significantly different at any time point in the BRX90 group. Of note, at Day 7, there was a high placebo response, with a higher proportion of patients in the placebo group than in the BRX90 group achieving remission. The proportion of patients achieving remission in the BRX60 group at Day 7 was only slightly higher than in the placebo group at this timepoint.
In terms of response, the proportion of patients with a response to both BRX60 and BRX90 was higher than in the placebo group, beginning at 24 hours and this difference was sustained through to Day 30. This difference was statistically significant at 24 hours, 36 hours, 60 hours, 72 hours and Day 30 in the BRX60 group, and at 60 hours and Day 30 in the BRX90 group. The change from baseline in MADRS was also statistically significant in the BRX60 group at both the 60-hour (p=0.0054) and 30-day (p=0.0447) timepoints (Table 1).
A total of 108 women were enrolled in Study 202C and were randomized 1:1 to receive placebo (n=54) or brexanolone 90 µg/kg/h (n=54). After 60 hours of infusion, the mean reduction in the HAMD-17 total score was 14.6 points in the BRX90 group and 12.1 points in the placebo group (p=0.0160). In terms of remission, the proportion of patients in the BRX90 group who achieved remission was numerically higher at 8 hours, 12 hours, 24 hours, 36 hours, 48 hours, 60 hours, 72 hours and Day 7. This difference was statistically significant at 48 hours, 60 hours, 72 hours and Day 7, but was not statistically significant at Day 30. At Day 30, the placebo remission response exceeded the BRX90 remission response.
In terms of response (Table 1), the proportion of patients with a >50% reduction in HAMD-17 total score was numerically higher in the BRX90 group at all timepoints measured from 24 hours to Day 7, with this difference achieving statistical significance at 48 hours, 60 hours, 72 hours and Day 7. The difference in percentage of responders was not statistically significant at Day 30, and in fact, placebo response rates exceeded the BRX90 response rates at this timepoint.
Brexanolone infusions were again found to be well tolerated in both studies, with the most common AEs including headache, dizziness and somnolence. Infusion site pain, nausea, dry mouth and fatigue were also reported as AEs. In study 202B, 51% of patients in the placebo group, 50% of patients in the BRX60 group and 54% of patients in the BRX90 group reported an AE. In study 202C, 45% of patients in the placebo group and 49% of patients in the BRX90 group reported an AE. Overall, 25 patients (18%) discontinued study 202B and eight patients (7%) discontinued study 202C. No patient discontinued the study because of AEs. Phase II and III trial outcomes are summarized in Table 1.
In 2019, brexanolone became the first drug specifically approved by the US Food and Drug Administration (FDA) for the treatment of PPD. Due to concerns about sedation and loss of consciousness, a Risk Evaluation and Mitigation Strategy of regular assessment of level of sedation and continuous monitoring of oxygen saturation using continuous pulse oximetry for the duration of the infusion are required. Recommended dosing is titration to 90 µg/kg/h over 60 hours, with observation for an additional 12 hours.
Zuranolone
Zuranolone is an investigational, oral NAS and GABA-A receptor PAM under investigation as a once-daily, 2-week therapy for depressive disorders.
Postpartum depression studies
ROBIN study Sage 217-PPD-201 (NCT02978326)
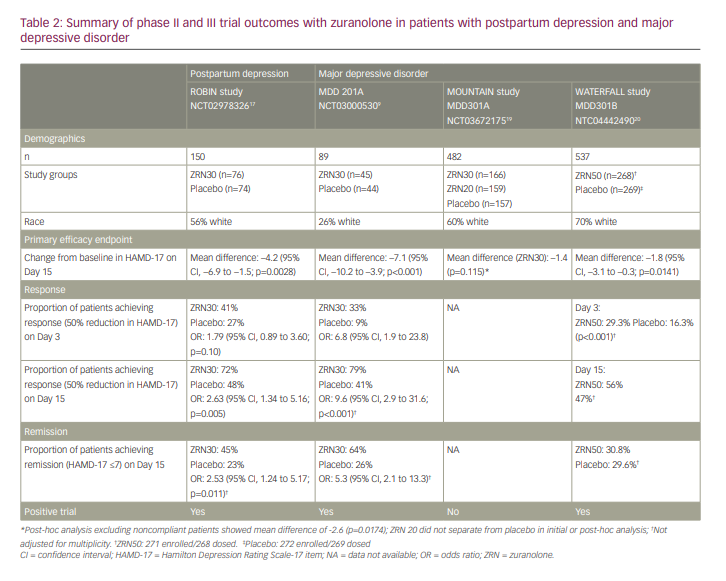
This double-blind, placebo-controlled, phase III clinical trial included 150 women aged 18–45 years who were ≤6 months postpartum, had onset of depression in their last trimester or within 4 weeks postpartum and had baseline HAMD-17 scores of >26.17 Seventy-six patients received 30 mg of zuranolone nightly for 14 days and 74 patients received placebo over the same period. Patients were allowed to be on an additional antidepressant but had to have been on a stable dose for more than 30 days and could not alter the regimen until at least Day 15 of the trial. Patients were assessed on Days 3, 8, 15, 21 and 45. The primary endpoint examined the change from baseline in HAMD-17 score at Day 15. Results showed statistically greater reduction in HAMD-17 total score with zuranolone compared with placebo at Day 15 (–17.8 versus –13.6, respectively; p=0.0028). Furthermore, zuranolone showed rapid reduction of symptoms as assessed by HAMD-17 total score by Day 3, with sustained superiority over placebo from Day 3 through to Day 45. Secondary endpoints included changes in MADRS score, Hamilton Anxiety Rating Scale (HAM-A) score, HAMD-17 response, HAMD-17 remission and Clinical Global Impression–Improvement (CGI-I) response. Outcomes for these measures supported the results of the primary endpoint. At Day 15, 72% of the zuranolone group showed HAMD-17 response versus 48% in the placebo group. Similarly, 45% in the zuranolone group achieved remission versus 23% in the placebo group at Day 15. Results at Day 15 for MADRS change from baseline and HAM-A improvement also favoured zuranolone over placebo. Zuranolone also continued to outperform placebo at Day 45 in all of these measures, with HAMD-17 remission at 53% compared with 30% with placebo, and response at 75% versus 57%, respectively.
In terms of AEs, approximately 60% in the treatment arm experienced at least one AE, compared with 52% in the placebo arm. The most common AEs in the treatment arm (>5%) were somnolence, headache, dizziness, upper respiratory tract infection, diarrhoea and sedation. The placebo group had similar common AEs, including headache, somnolence, nausea, dizziness, vomiting, abnormal dreams and hyperhidrosis. In both groups, most AEs were mild to moderate. An equal number in each arm (n=3) experienced severe treatment-emergent AEs and one patient from each arm experienced a serious AE. Three patients required dose reductions (to 20 mg) after experiencing sedation, confusion or somnolence. One patient discontinued zuranolone early due to intermittent sedation.17
An additional exploratory endpoint also examined remission using the EPDS, using three different values as remission definitions. At all three of these values (HAMD-17 scores <13, <10, <7), a significantly higher proportion of patients receiving zuranolone achieved remission compared with those receiving placebo.18 A similar study, SKYLARK (NCT04442503) examining zuranolone 50 mg versus placebo for PPD is under way, with results expected in late 2021.
Major depressive disorder studies
Study MDD 201A (NCT03000530)
Given the role of GABA in depression, zuranolone was further studied for efficacy in the treatment of MDD. The initial phase II trial was a double-blind study that included 89 patients who were randomized to receive either zuranolone 30 mg (n=45) or placebo (n=44) for 14 nights.9 All patients were aged 18–65 years and diagnosed with MDD with HAMD-17 score of 22 or above. They were either on a stable antidepressant dose for 30 days or more, or not on an additional antidepressant. Assessment frequency, and primary and secondary endpoints were similar to those in the ROBIN study, with the primary endpoint being the change from baseline on HAMD-17 total score at Day 15. In terms of the primary endpoint, the least-squares mean change from baseline on HAMD-17 was −17.4±1.3 points in the zuranolone group and −10.3±1.3 points in the placebo group (p<0.001). Secondary endpoints were similarly promising, with zuranolone outperforming placebo in most measures (Table 2). There were no serious AEs in either group, though two zuranolone patients had AEs that met criteria for discontinuation according to the protocol (one patient with nausea, dizziness and headache, and the other with elevated liver enzymes, which normalized after study discontinuation). The most common AEs (>5%) in the treatment arm were headache, dizziness, nausea and somnolence. AEs were somewhat more likely in those receiving zuranolone while on an additional antidepressant than those on zuranolone monotherapy (67% versus 48%). In addition to the two discontinuations, six patients required dose reductions due to AEs.9
MOUNTAIN study (MDD301A) (NCT03672175)
Following the promising phase II trial, this phase III trial examined oral zuranolone 30 mg (n=166), zuranolone 20 mg (n=159) and placebo (n=157) taken for 14 nights. Patients were seen in follow-up visits from Day 3 to Day 42, and then with monthly naturalistic follow-up to 6 months. Tolerability was similar to phase II data, with patients reporting similar AEs. Zuranolone 20 mg failed to separate from placebo at any measured timepoint, including the primary endpoint of Day 15 (Table 2). Zuranolone 30 mg achieved statistically significant improvement/differences at Days 3, 6, 9 and 12 but failed to separate from placebo at the primary endpoint of Day 15 (change from baseline –12.6 versus –11.2 points, respectively; p=0.115). Post-hoc analysis indicated that approximately 9% of the patients in the zuranolone 30 mg group had no detectable drug level, indicating non-compliance. When the data were reanalysed after excluding these patients, statistical significance was achieved at the primary endpoint.19
Given the inconclusive results of the MOUNTAIN study, and to fully understand the efficacy and safety profile of zuranolone, additional studies are now under way to assess the efficacy of zuranolone in MDD.
WATERFALL study (MDD-301B) (NCT04442490)
The WATERFALL study20 examined the higher dose of zuranolone 50 mg versus placebo given daily for 14 nights with or without a conventional daily oral antidepressant with a primary endpoint of change from baseline in HAMD-17 at Day 15, which mirrors the MOUNTAIN study through to Day 42. This phase III trial compared zuranolone 50 mg (n=271) or placebo (n=269) taken for 14 nights. Zuranolone 50 mg was statistically superior to placebo at Day 15 (primary endpoint), following the last dose of the 2-week treatment period (change from baseline in HAMD-17 total score, –14.2 versus –12.3 points, respectively; p=0.0141) (Table 2). Differences were also nominally significant (p<0.001) for HAMD-17 total score at Day 3, 8 and 12. AEs were consistent with the safety profile in previous studies (somnolence, dizziness, headache and sedation seen in >5% of zuranolone treatment group) and were mild to moderate in severity. No tolerability differences were seen in patients continuing on previous standard oral antidepressant therapy plus zuranolone versus zuranolone monotherapy. Responders to zuranolone 50 mg at Day 15 retained 86% of their HAMD-17 response at Day 42 (4 weeks after cessation of study treatment).20,21
SHORELINE study (MDD-303) (NCT03864614)
The SHORELINE study is an open-label, longitudinal study examining the need for re-treatment with zuranolone through 1 year, as well as safety and tolerability. After initial response to zuranolone 30 mg for 14 days, patients were assessed every 2 weeks and those with PHQ-9 scores ≥10 and HAMD-17 ≥20 were re-treated. A minimum of 56 days between treatment cycles (each cycle lasted 14 days) was required to allow for a maximum of five 14-day courses of zuranolone 30 mg in 1 year. The trial is on-going but an interim analysis has shown promising results. Patients with MDD (HAMD >20 and MADRS >28) received zuranolone 30 mg (n=725); 304 of them were on concurrent antidepressant therapy while 421 were treated with zuranolone alone. Patients who did not respond to the initial 14-day treatment course exited the study (173 patients). Among those who had a response to initial treatment (489 patients), 210 (42.9%) did not require any re-treatment throughout the following year. An additional 125 patients (25.6%) required only one re-treatment cycle. Thus, nearly 70% of study patients needed only 1–2 treatment cycles over 1 year. Only 154 patients (31.5%) required more than two treatment cycles. Furthermore, change in baseline in HAMD-17 total score at Day 15 was similar between the zuranolone and zuranolone plus standard antidepressant groups in each treatment cycle, as was need for re-treatment and reported AEs.22 Again, given results of the MOUNTAIN study, it is likely that the 30 mg dose of zuranolone is the minimal effective dose. Thus, the dose of zuranolone in the SHORELINE study was changed to 50 mg with re-treatments for patients who received 30 mg as the first treatment, and starting with 50 mg in initial treatment for new patients. Data collection continues.
An interesting additional study is the CORAL study (NCT04476030), which is examining zuranolone as a rapid response treatment to be used concurrently with SSRI initiation. Enrolment is ongoing with zuranolone 50 mg versus placebo, each co-initiated with open-label antidepressant in patients with MDD.23
Ganaxolone
Like zuranolone, ganaxolone is a synthetic analogue of the NAS allopregnanolone. Together, brexanolone, zuranolone and ganaxolone (as well as experimental drug Prax-114) constitute a class of novel GABA-A receptor selective PAM antidepressants. Given prior studies demonstrating an inverse association between depression symptom severity and allopregnanolone serum levels, as well as decreased levels of the precursor to allopregnanolone, progesterone in postmenopausal women, an open-label pilot study was conducted to study ganaxolone in 10 postmenopausal women with MDD refractory to adequate antidepressant treatment (NCT02900092).24 The study evaluated changes in MADRS scores over the 8-week period of treatment. Nine out of 10 women completed the 8-week treatment. Four of the nine demonstrated a response to treatment, defined as a 50% reduction in baseline MADRS score, as well as remission, defined as a final MADRS score <10. There were also significant improvements in the secondary endpoints evaluating individual mood symptoms including anxiety, suicidal ideation, sleep disruptions and appetite changes. There was no significant worsening of symptoms on any primary or secondary endpoints; however, all patients experienced sleepiness and fatigue, and six patients experienced dizziness. Finally, improvement in MADRS scores was maintained after a 2-week drug taper at Week 10.
Additionally, currently registered trials for ganaxolone include two phase II, double-blind, placebo-controlled, studies to assess safety and efficacy of (1) intravenous ganaxolone (NCT03228394),25 and (2) oral ganaxolone (NCT03460756)26 in PPD. These trials completed in 2020, but no results have yet been released. Moreover, a phase II trial (NCT01339689) assessing safety and efficacy of ganaxolone in post-traumatic stress disorder (PTSD) failed to separate from placebo. Under-dosing may have contributed to this outcome.27
PRAX-114
An additional orally administered GABA-A receptor PAM, which has yet to be named and is known as PRAX-114, is currently being assessed for safety and efficacy as monotherapy for the treatment of severe MDD (Study PRAX-114-213, NCT04832425).28 The phase II/III study was initiated in March 2021, and is a randomized, double-blind, placebo-controlled clinical trial of 200 patients receiving either 40 mg of PRAX-114 or placebo daily for 28 days. Enrolment is continuing.
Praxis has also announced plans to initiate two phase II studies to assess PRAX-114 for (1) adjunctive treatment to standard antidepressant therapy in MDD (Study PRAX-114-214, NCT04969510), and (2) as a treatment for PTSD. Both studies are expected to start in late 2021.
Discussion
Brexanolone
The results from the clinical trials of brexanolone are promising, with all three trials meeting their primary endpoints. Brexanolone works quickly, with a reduction in HAMD-17 total score observed within 60 hours after the infusion was started in all study groups, which is a meaningful advantage when compared with conventional antidepressant treatment. Remission was observed from 48 hours after the start of treatment in both the BRX60 group in study 202B and the BRX90 group in study 202C. The long time to oral antidepressant action has been a problem in the field for decades; brexanolone holds promise for an agent that acts rapidly compared with conventional antidepressants.
In a meta-analysis using matching-adjusted indirect comparisons to evaluate the efficacy of brexanolone infusions versus SSRIs for treatment of PPD, there was a greater change from baseline in both the HAMD-17 and EPDS scores in patients who received BRX90 compared with SSRIs and the difference was statistically significant after Day 3 of initiating treatment.29 At 4 weeks following treatment initiation and at last observation, change from baseline in HAMD-17 and EPDS scores was generally equivalent or slightly better (though not statistically significant) with BRX90 compared with SSRIs. BRX90 was used in this meta-analysis because there are more data available for BRX90 than for BRX60.
It is also worth noting that studies 202B and 202C enrolled a diverse patient population that is representative of the US population. In study 202B, 66% of patients were white and in study 202C, 60% of patients were white. In study 202A, 38% of patients were white. There was no difference in outcomes in women taking oral antidepressants versus those not taking an oral antidepressant in these trials, meaning that the antidepressant mechanism of action of brexanolone is unique and likely does not involve direct effects on the monoamines.
Interestingly, although all studies met their primary endpoints, both remission and response data in study 202B seem to favour BRX60 over BRX90 in treatment of severe depression. This would suggest that clinically, the best approach to treating severe depressive episodes in the postpartum setting is to use the BRX60 dose. Given that there was no difference between the groups of women who were taking and not taking an antidepressant, it may not be necessary to start an antidepressant when brexanolone treatment is initiated. One limitation of these studies is that patients were only followed up to Day 30. Thus, responders to brexanolone should be monitored carefully and a standard-of-care treatment should be considered if depressive symptoms return/increase. Appropriate standard-of-care antidepressant therapy should be considered/offered to women who have not responded by the end of the infusion.
Brexanolone has also shown efficacy in the treatment of moderate depression, with both remission and response being achieved at 48 hours and lasting up to Day 7. Only the BRX90 dose was studied in the trial with women who have moderate depression and it would be interesting to compare BRX60 with the BRX90 dose given the apparent superior efficacy of BRX60 in severe depression. Brexanolone was well tolerated overall; the most common AEs included drowsiness, presyncope, dizziness and vertigo. The FDA issued a boxed warning to inform patients of the risk of excessive sedation and sudden loss of consciousness. Because of this warning, patients receiving brexanolone need continuous pulse oximetry monitoring throughout the infusion, and brexanolone is only available for administration through the Zulresso Risk Evaluation and Mitigation Strategy programme.30
Future studies comparing outcomes of BRX60 versus BRX90 at timepoints beyond 30 days after the infusion would also be useful to inform dosing choices. Data past 30 days in women on oral antidepressant therapy versus not on oral antidepressant therapy would also help guide the clinical decision on whether or not it would be useful to start an antidepressant at the time of brexanolone infusion.
Zuranolone
As with brexanolone, zuranolone has shown promise in the treatment of PPD. In addition to meeting its primary endpoint, the rapid decrease in depressive symptoms and sustained improvement at Day 45 in the ROBIN trial bolsters the potential clinical significance of this medication. The exploratory endpoint examining reduction in the EPDS may be especially useful given the widespread use of this scale by paediatricians, obstetricians and psychiatrists in the postpartum period. The results indicate that zuranolone 30 mg can be of use in those with severe PPD, and offers an alternative to patients who are unable or unwilling to be admitted for brexanolone administration. Furthermore, the results show zuranolone to be a safe and well-tolerated medication, with largely similar AEs as placebo and only one patient who required discontinuation.
The results for MDD are more complex, but ultimately still promising. Phase II data showed rapid and significant improvement with zuranolone compared with placebo using many different measures. Though the initial phase III trial of zuranolone 30 mg did not meet the primary endpoint, post-hoc analysis indicated significant non-compliance and with these patients removed, the data showed similar results to the phase II trial. This study does indicate that the minimum effective dose is probably 30 mg, and the 50 mg trial was positive. Additional trials will help to indicate whether 50 mg leads to greater efficacy and/or a higher response rate. Initial data from the SHORELINE study shows that a significant number of patients who initially respond to zuranolone sustain that improvement at 1 year without the need for additional treatment, even among those not on an additional antidepressant. The implications of this are significant, as this may lead to a number of patients with sustained improvement for over 1 year after only one 2-week treatment period. These patients can avoid the burden, including side effects and cost, of taking a daily antidepressant. Theoretically, the subgroup of patients with MDD who have low levels of GABA in the cerebrospinal fluid and plasma will respond particularly well to zuranolone and other GABA-A PAMs. Testing for GABA levels is not done routinely but may prove to be helpful in predicting who will respond to these types of medications.
A potential further benefit of zuranolone compared with standard antidepressants comes from examining sexual dysfunction, a well-recognized adverse effect seen with administration of most antidepressants currently available. This adverse effect typically emerges 1–3 weeks after treatment initiation,31 with prevalence as high as 73% depending on the antidepressant administered.32,33
An exploratory post-hoc analysis of the MOUNTAIN trial participants examined sexual dysfunction using the Changes in Sexual Functioning Questionnaire-14 (CSFQ-14) at baseline and at Days 15, 28 and 42.34 There was no difference between zuranolone 30 mg and placebo in sexual functioning after treatment initiation for either men or women as measured by CSFQ-14 total score. Subscale score results were similar, with no phase-specific signal for either women or men. Additionally, both groups had similar improvement among those who had pre-existing impairment due to depression, with sexual dysfunction declining from 82.4% at baseline to 66.5% at Day 42.34
GABA-A receptor PAMs are a promising and attractive option for treatment of MDD, particularly for patients with peripartum/perimenopausal onset or worsening. Extrasynaptic (tonic) inhibition at the GABA-A receptor13 has been implicated in the long-lasting antidepressant effects of these agents in trials to date. Both brexanolone and zuranolone demonstrated early and sustained benefit in treatment of women with PPD. After a promising phase II trial assessing zuranolone in patients with MDD, a phase III study failed to demonstrate significant benefit at the primary endpoint (Day 15) for the zuranolone 30 mg dose, although post-hoc analysis identified no detectable drug level in a portion of the patients, which impacted findings. Given the results of this study, a higher dose of 50 mg of zuranolone was added to assess the effectiveness for treatment in MDD versus placebo. Additionally, ganaxolone demonstrated a clinically significant benefit over placebo for treatment of MDD in a small study of postmenopausal women. The most common AEs in these studies were sedation and dizziness, making night-time administration most appropriate.
Conclusion
In summary, initial studies have shown promising results by demonstrating early and sustained benefit of NAS GABA-A receptor PAMs for treatment of MDD in women and men. With studies demonstrating onset of improvement much earlier than any currently available treatments, short duration of treatment course, good tolerability and long-term maintenance of effect, these agents have the potential to drastically change the trajectory of illness and quality of life of patients with depression.







