Cerebral amyloid angiopathy (CAA) is a disorder of the central nervous system characterised by the deposition of amyloid proteins in the wall of small- to medium-sized vessels, most frequently arteries, within the leptomeninges and cortex of the brain.1 In vessels affected by CAA, local muscle and elastic elements of the arterial wall are lost and replaced by amyloid fibrils, primarily the amyloid-β (Aβ) peptide. Since the first description of neurovascular amyloid deposition in 1909 by Gustav Oppenheim, sound scientific evidence has supported the concept that the associated disruption of the overall structure of those small vessels predisposes to both ischaemic small vesseldisease and cerebral haemorrhage.2–4
Sporadic CAA is a major cause of lobar intracerebral haemorrhage (ICH) and cognitive decline in the elderly, including the normotensive population.5,6 Hereditary forms of CAA are generally rare, usually more severe and earlier in onset. Rare non-Aβ familial CAAs can also present clinically with lobar ICH.7 Regarding sporadic CAA, two major challenges persist:
- a definitive diagnosis requires a neuropathological exam; and8
- no treatment or preventive strategy for CAA or CAA-ICH has been firmly established.
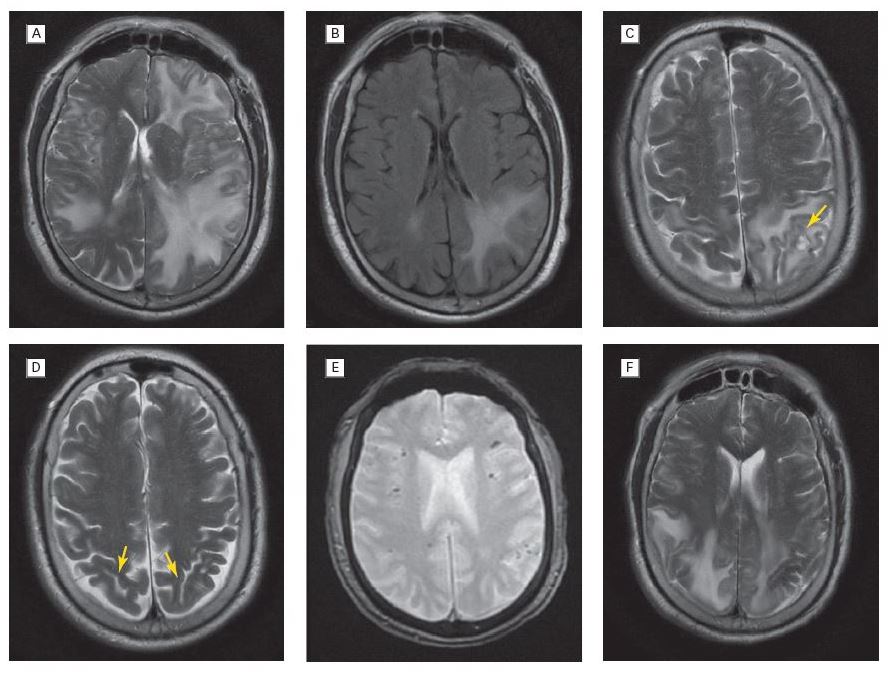
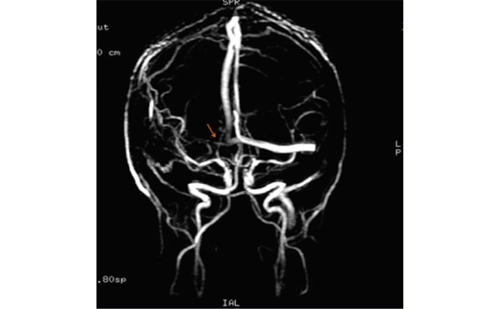
Nevertheless, in the last decades of research, there has been remarkable progress in our understanding of this condition. CAA pathology has been associated with markers of small vessel disease, including lobar cerebral microbleeds (CMB) and white matter hyperintensities on magnetic resonance imaging (MRI).9–11 The availability of MRI sequences that are particularly sensitive to susceptibility effects like the T2* gradient-recalled echo (GRE) and susceptibility weighted imaging (SWI) now allow reliable assessment of an individual’s haemorrhagic burden over time and reasonable accuracy by clinical and neuroimaging diagnostic criteria.10–13 As our understanding of CAA pathophysiology evolves, specific targets have been identified as candidates for the prevention and treatment of this condition.14 As newresearch tools such as the Pittsburgh Compound B (PiB) or other amyloid-imaging agents for positron emission tomography (PET) scan become incorporated into clinical practice, it may also be possible to detect vascular amyloid deposition in the brain noninvasively in living patients, perhaps before an ICH or significant cognitive decline.15
To view the full article in PDF or eBook formats, please click on the icons above.