Cognitive impairment (CI) is frequently associated with critical illness and can be defined as the loss or decline of higher mental functions (memory, attention, calculation, language, orientation and speed of information processing) that modify a person’s activity and social interaction. CI is additionally defined as self- and/or informant-reported involving decreased ability on cognitive tasks and/or preserved basic activities of daily living/minimal impairment in complex instrumental functions. Decline in cognitive functions can have an evolving course. The patient has the ability to perform daily living activities, except those that require complex cognitive instruments. Long-term cognitive impairment after critical illness (CIACI) is an emerging medical concept that was first described in 1999 in a report of a cohort of 55 patients with acute respiratory distress syndrome (ARDS) among whom 78 % had CI (decreased memory, attention and concentration or lower mental speed) at 1 year.1
This article aims to outline the current knowledge of CIACI and to present a hypothesis of novel treatment to prevent CIACI based on relevant research and clinical data. The authors overviewed the major clinical features of CIACI. We have also outlined the probable underlying pathological mechanisms as well as endogenous biological defence processes that are the potential targets for novel treatments. Finally, we overviewed current therapeutic approaches as well as proposed a new treatment strategy based on the hypothesis that an intervention at the neurotrophic regulation level could potentially address both neuroprotection and neurorepair in the critical care period and beyond, counteracting and/or preventing development of CIACI. The recently published data indicating that delirium is an independent risk factor of CIACI2 suggests that this group of patients might benefit from timely administered preventive therapies.
Major Clinical Features of CIACI Incidence
In various studies, CI is has been reported as a frequent consequence of critical illness.3 One example was a large prospective cohort study of 821 patients with respiratory failure or shock, conducted in the US.1

The occurrence of delirium during hospitalisation was assessed by the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU) and overall cognition and executive function at 3 and 12 months after discharge using the cognitive test battery RBANS (Repeatable Battery for the Assessment of Neuropsychological Status) and Part B of the Trail Making Test, respectively. This revealed that 66 % of patients had CI at 3 months. Among these, 40 % had global cognition scores compatible with mild CI and 26 % had scores well matched with mild dementia. Deficit occurred in both older and younger patients and persisted, with rates of 34 % and 24 % on assessments at 12 months.
Underlying Pathology
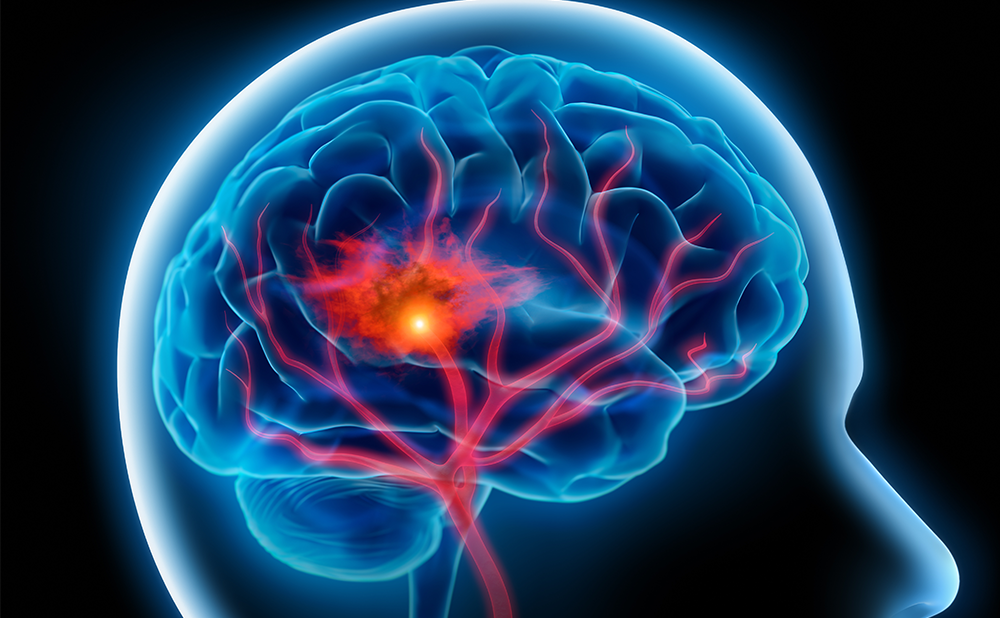
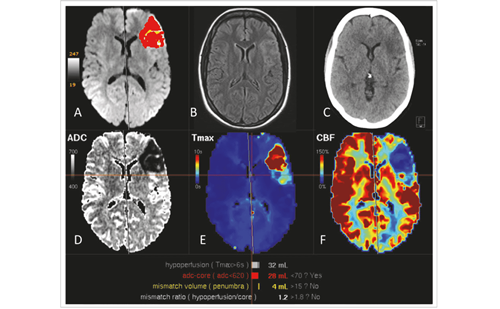
There is an incomplete knowledge available regarding underlying pathological events in the development of CIACI. Here, only a few important aspects of this complex topic are highlighted. Studies of brain pathology in critical illness have focused mainly on sepsis-associated observations.4,5 Post-mortem studies in people who had septic shock demonstrate cerebral oedema, ischaemic lesions, haemorrhage, microthrombi, microabscesses and leukoencephalopathy (see Figure 1).6,7 These abnormalities point to and overlap with those described after stroke and traumatic brain injuries and in various neurodegenerative disorders.8–11 A significant apoptosis is also observed in central nervous system (CNS) sectors with activity related to autonomic cardiovascular function.6,7 These findings suggest the existence of another underlying mechanism of circulatory collapse in sepsis probably mediated by brain damage.
Animal models using lipopolysaccharide challenge, proinflammatory cytokine administration or caecal ligation provided evidence of altered blood–brain barrier (BBB) function and entry of inflammatory mediators into the CNS. They also showed activation of brain innate and adaptive immune systems12 and induction of neuronal and glial dysfunction and death.13 These processes are accompanied by the expression of toll-like receptors and tumour necrosis factor (TNF) receptor-1, which are of major importance to amplifying peripheral inflammatory signals within the brain.14
Brain Imaging
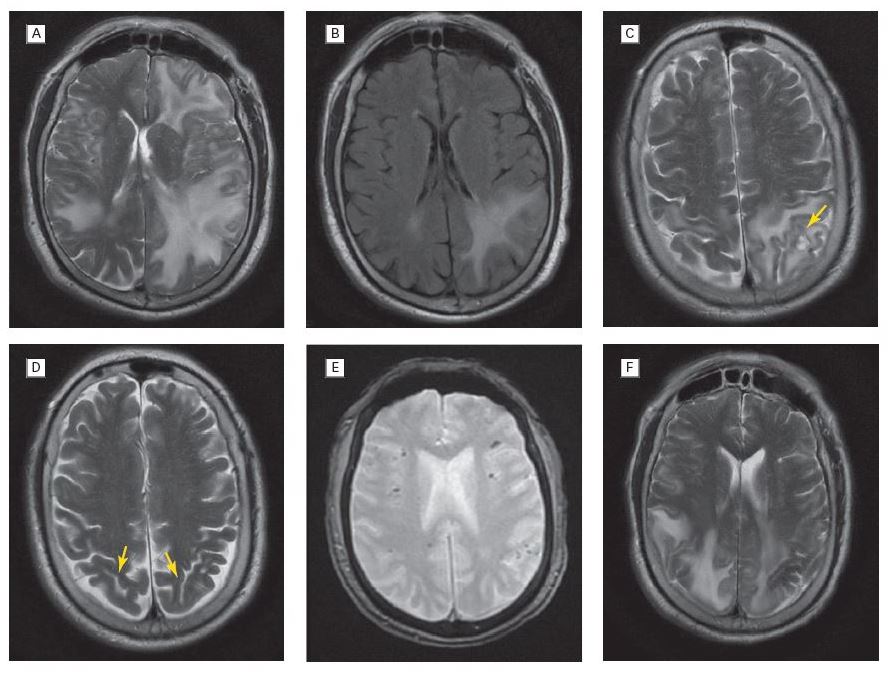
Recently, the lack of neurological images of patient populations in ICUs has been addressed in several clinical studies. One of these, the VISualizing ICU SurvivOrs Neuroradiological Sequelae (VISIONS) study was a prospective observational trial that analysed the association between duration of delirium, brain volumes and CIACI.15 Patients with a longer duration of delirium showed greater brain atrophy at hospital discharge and 3 months follow-up and were associated with worse cognitive performance at 12 months. Smaller superior frontal lobes, thalamus and cerebellar volumes at 3 months were associated with worse executive functioning and visual attention at 12 months (see Figure 2). Longer durations of delirium were also associated with disruption of white matter in the knee and in the splenium of the corpus callosum and the anterior end of the internal capsule at discharge (see Figure 3). Lower fractional anisotropy in the front end of the internal capsule at discharge and corpus callosum knee at 3 months were associated with poorer cognitive scores.16
In another study, the structural changes in brain imaging and cognitive outcomes were retrospectively evaluated in a cohort of 64 critically ill patients who developed neurological changes during their ICU stay.17 CIACI occurred in 48 % of survivors who had abnormalities on X-ray computed tomography (CT) and in 64 % of them with lesions detected on magnetic resonance imaging (MRI). Lesion location was heterogeneous. The study included 19 patients with white matter hyperintensities, which the American Heart Association believes should be interpreted as silent infarcts.18

Patient-associated Risk Factors of CIACI Depression
Some data suggest that depression in the post-ICU period is associated with CI.19–23 This was emphasised in a cross-sectional study of 102 long-term survivors of ARDS, in which 66 % had psychiatric symptoms (depression, anxiety or post-traumatic stress disorder) and concomitant CI was significantly associated with these symptoms (p=0.04).24 An analysis of 109 survivors of ARDS at 3, 6 and 12 months after ICU discharge using the 36-item Short-Form General Health Survey (SF-36) showed that scores for general health, vitality, social functioning and mental illness were low and remained low at 1 year.25 Longer duration of mechanical ventilation and delayed recovery of organ failures predicted worse Beck Depression Inventory (BDI-II) score in the following 5 years.26,27 A systematic review of 14 studies on depression in critical-illness survivors found that one out of three patients had moderate to severe symptoms of depression and early onset of depression at discharge, and there was a high risk of later depressive symptoms.28
Apolipoprotein E4 and Other Markers of Alzheimer’s Disease
The detection of biomarkers used in Alzheimer’s disease (AD), such as apolipoprotein E (APOE), may be helpful in identifying patients with increased risk of cognitive changes after surgery and hospitalisation in the ICU.29 One study found that the presence of APOE4 had a stronger association with delirium duration than age, severity of illness, sepsis or use of benzodiazepines30 although this association was not supported by other studies.2
Independent Risk Factors of CIACI Delirium
Delirium is an acute change in mental status with fluctuating course characterised by inattention, disorganised thinking or altered states of consciousness. It is very common in acute disease and has been reported in 74 % of patients with a critical condition. The longer duration of delirium is an independent risk factor for CI and impaired executive function at 3 and 12 months after discharge from ICU.2
To date, no single cause of delirium has been identified. Known risk factors include advanced age, pre-existing CI, drugs (especially benzodiazepines and anticholinergics), sleep deprivation, hypoxia, metabolic abnormalities and history of alcohol or drug abuse. Several theories have been proposed to explain the development of delirium, most of which are complementary (see Table 1).31
Drug Exposure
ICU patients are likely to receive multiple pharmacological agents, many of which can interfere with nerve function. Benzodiazepines (mainly midazolam and lorazepam), for example, are widely used for sedation in those receiving mechanical ventilation. A cohort study of 198 patients with mechanical ventilation found that lorazepam was an independent risk factor for delirium. Fentanyl, morphine and propofol were associated with an increased but non-significant risk of delirium. In addition, increasing age and Acute Physiology and Chronic Health Evaluation II (APACHE II) scores were also independent predictors in the transition to delirium (p<0.05).32 The BRAIN-ICU Study,2 however, showed that use of sedatives or analgesics was not consistently associated with CI at 3 and 12 months. Drugs with potential central anticholinergic effects (tricyclic antidepressants, H2 blockers, opiates, furosemide and benzodiazepines) may affect neurotransmission and cause delirium but have been reported to affect only short-term outcomes in mechanically ventilated ICU patients.33–36
Current Protocols for Prevention and Treatment of CIACI
Pain, Agitation and Delirium Prevention
Strategies for pain, agitation and delirium prevention in the ICU with sedation suspension protocols, spontaneous ventilation assays, early mobility and sleep hygiene programmes are associated with significant improvements in performance and reduced cost of care. This ‘delirium care bundle’ is detailed in the Society of Critical Care Medicine’s 2013 guidelines.37
Physical and Mental Rehabilitation
In patients with dementia, exercise is associated with increased cerebral blood flow, neurogenesis and brain volume, which could be linked with improved cognitive level. These effects are also shown in severe head trauma survivors, based on the principles of neurological compensation or neurological restoration. Such findings suggest that the human brain recovers from injury to differing extents.38
A review performed by the Institute of Medicine and Emergency Care showed that cognitive rehabilitation therapy is effective for improving some of the deficits associated with traumatic brain injury (TBI), but overall the evidence was insufficient to determine the full therapeutic value of this therapy.39 Two small but conclusive trials showed the importance of cognitive rehabilitation in critical care. The Returning to Everyday Tasks Utilising Rehabilitation Networks (RETURN) study40 was a singlesite, randomised trial of 21 general medical/surgical ICU survivors (eight controls and 13 intervention patients) with either cognitive or functional impairment at hospital discharge. At 3-month follow-up, patients who received in-home cognitive, physical and functional rehabilitation demonstrated significantly improved cognitive executive functioning compared with controls who received standard care. A multicomponent rehabilitation programme for ICU survivors, therefore, may be an effective approach to improving cognitive performance and functional outcomes.

In the Activity and Cognitive Therapy in ICU study,87 medical and surgical ICU patients with respiratory failure and/or shock were randomised in a 1:1:2 ratio to usual care, early once-daily physical therapy or early once-daily physical therapy plus a novel, progressive, twice-daily cognitive therapy protocol. Functional- and health-related quality-of-life outcomes did not differ between groups at 3-month follow-up, but the study demonstrated that early rehabilitation can be extended beyond physical therapy to include cognitive therapy.41
Key Prerequisites for Development of Novel Therapies for CIACI
CIACI, together with the age-related dementias and cognitive disorders
linked to the plethora of other aetiological factors, represents an
increasing challenge to medicine and the society. In spite of a long history
of failure in clinical trials, especially in the so-called neuroprotection
domain, there is a growing understanding of key requirements that
need to be fulfilled for development of new therapies in neurology.
Here, we attempt to outline some of these prerequisites:
- Definition of biologically accurate treatment targets.
- Identification of suitable/adequate therapeutic agents and regimens.
- Patient stratification and sound clinical trial design.
Neurovascular Unit as Biologically Relevant Treatment Target
Several pathological features and mechanisms preceding or accompanying CIACI match those identified in other neurological disorders. Accordingly, some of the potential treatment targets can be considered the same or similar to those described in vascular dementia, TBI and stroke (e.g. prevention of programmed cell death, apoptosis). However, we believe that the effective novel therapies must target biologically relevant structures and processes, not isolated pathological events (e.g. excitotoxicity). The main structural and functional constituents of the brain are represented by the concept of neurovascular unit (NVU), and the focus of future therapies should be on the protection of NVU as well as on ability to stimulate its restoration.
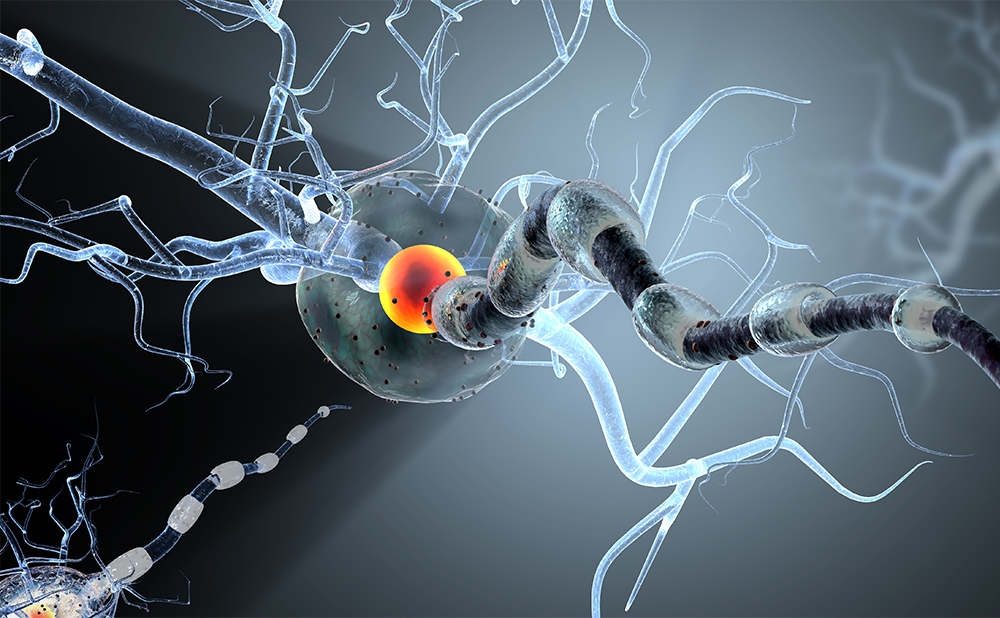
The ‘neurovascular unit’ is a concept linking microvessel and neuron function and the responses of these compartments to injury.42 The NVU is a dynamic structure assembled by endothelial cells enclosed by a basal lamina, and is surrounded by astrocytic end-feet processes, pericytes and neurons (see Figure 4).43–46 A key function of the NVU is to regulate plasma components and cellular elements of brain intravascular space passage. This barrier function, also known as the BBB, is determined not only by endothelial integrity but more specifically by a functional interaction between endothelial cells, basal lamina and perivascular astrocytes.47 Endothelial permeability is restricted by junctional complexes gathered by adherents and tight junction (TJ) proteins. A set of accessory proteins, known as zonula occludens, links these proteins with the cytoskeleton.48 At the NVU, it is primarily TJ proteins that confer the low paracellular permeability and high electrical resistance that characterise the cerebrovascular endothelium. The NVU is prominent throughout the brain, including the grey and white matter. NVU dysfunction is involved in several brain conditions, such as stroke, vascular dementia, migraine, trauma, all neurodegenerative disorders and even normal ageing.48 As discussed earlier, critical illness can lead to similar pathological features. Therefore, the maintenance and restoration of NVU functions also represent proper targets for potential therapies to prevent CIACI.
The Mechanisms of Cell Death and Brain Repair are Linked
Brain cell generation, function and death involve four key concepts. Neurotrophicity describes cell processes aimed at maintaining correct DNA expression and cell/tissue phenotype. Neuroprotection is a shortor medium-term endogenous neurobiological process that includes all the mechanisms directed against harmful factors. Neuroplasticity describes the brain’s ability to change existent structures in response to environmental stimuli, such as learning, new experiences or injury. Neurogenesis is the process by which new nervous tissue cells, such as neurons, astrocytes and oligodendrocytes, are formed from stem cells.49,50 These processes are simultaneously regulated and integrated in both the healthy and diseased brain and can be endogenously or pharmacologically activated, allowing patients to overcome insults and stimulate recovery.51
The results of reduced neuroplasticity include neuropathic pain, multiple sclerosis, tinnitus, movement disorders and obsessive compulsive disorder.52 Changes in neurogenesis can also generate pathological conditions such as AD (downregulation) and neuroproliferative disorders (upregulation).
Excitotoxicity results from excess of glutamate or similar substances. When N-methyl-D-aspartate receptors (NMDAR) are stimulated at low intensity by calcium, they participate in proteolitic systems, affecting neurotrophism and neuroplasticity.50,53 When activated in excess (as after an insult), however, they have a destructive role.
Inflammation regulates these processes and therefore affects both neuroprotection and neuroplasticity. Therefore, NMDAR can have both protective and damaging roles in pathological processes. Apoptosis is a positive process (only when controlled), but apoptosis-like processes (uncontrolled apoptosis) induced by overactivation of NMDAR are always negative and must be addressed in therapeutic interventions.54
We know that in the CNS, all these processes are regulated partly by neurotrophic factors (NTFs). Their positive or negative impact on NVU depends on the balance between endogenous defence activities (EDA) and damage mechanisms (DM) (see Table 2), which in turn depend on neurotrophic regulation.50 These considerations are important for defining properties/pharmacological profiles of potential therapeutic agents.
NTFs as Therapeutic Agents in CIACI
In 1951, Cohen, Levi-Montalcini and Hamburger demonstrated that mouse sarcoma could stimulate sympathetic and sensory neuron growth, and they isolated nerve growth factor (NGF).55
NGF was shown to interfere with oligodendrocyte migration in the CNS and with migration of Shwann cells in the peripheral nervous system. This led to a closer investigation of specific nervous-system cytokines (brain-derived NTF [BDNF], glial-derived NTF [GDNF]), and the investigators were awarded the Nobel Prize for this research in 1986.56 The development of neurotrophic treatment concept has been well described by Aloe et al. 2012 (see Figure 5).57
Neurotrophins (NGF, BDNF, neurotrophin-3 [NT-3] and neurotrophin 4 [NT- 4]) are of interest due to their involvement in the normal development of the CNS and in the normal or pathological ageing.58 These factors exert their effect through tropomyosin kinase (TRK)-related receptors and activation of signalling cascades including Inositol triphosphatediacylglycerol (IP3-DAG), Phosphatidylinositol-3 kinase (PI3K)/Akt and mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK). They also interact, with low affinity, with p75NTR, a TNF receptor, which, upon activation, leads to apoptosis in neuronal and non-neuronal cells.58
The best-studied example to date is BDNF, which was found to be involved in almost all stages of development of neural circuits including:59–61
- stem cells’ and progenitors’ survival;
- neurogenesis and neuronal differentiation;
- polarisation and neuronal guidance;
- branching and survival of differentiated neurons; and
- formation and maturation of spines and synapses.
In the mature nervous system, BDNF promotes development and refinement of neuronal circuit structure, modulates synaptic plasticity and therefore regulates cognitive brain functions (including learning and memory). Alterations in BDNF levels are associated with neurodegenerative disorders (including AD, Huntington’s disease and epilepsy), neuropsychiatric disorders (including depression, anxiety disorders, bipolar disorders, schizophrenia and addiction) and obesity.62–65 The hallmark of BDNF deficiency is synaptic degeneration, and this is consistent with the finding that increased levels of BDNF promote synaptic repair in preclinical models. BDNF has a major role in regulating survival, growth and maintenance of neurons and has an important role in learning and memory processes.66,67 Low levels of BDNF have been found in patients with AD and major depression and

are considered a biomarker of memory disorders, cognitive function and increased risk of death in elderly women. BDNF is also decreased in type II diabetes. Vigorous exercise increases muscle levels of BDNF as well as its cerebrospinal fluid (CSF) levels, showing an autocrine and, perhaps, paracrine effect. There is a clear relationship between elevated levels of interleukin 6 (IL6), BDNF, exercise and improved cognitive function.66–69 BDNF is a highly charged protein that does not readily cross the BBB, so effective delivery to the CNS is a challenge.70
GDNF has neuroprotective effects in dopaminergic neuron-cell cultures. Mesencephalic astrocyte-derived NTF and conserved dopamine NTF are members of an NTF family with specific protective properties on dopaminergic neurons. This was shown in 6-hydroxydopamine animal models of Parkinson’s disease.71 The family of insulin-like peptides (insulin growth factors [IGFs] 1 and 2) are generated within the brain or enter the brain through the BBB attached to a low-density lipoproteinrelated protein 1 (LRP1) receptor.72 They concentrate in the hippocampus and hypothalamus, exerting regulatory actions on neuronal survival, neurogenesis, angiogenesis, inhibitory or excitatory neurotransmission and cognition. The tissue level of IGF 1 is therefore an important element of healthy nervous tissue; aberrant reduction of IGF 1 may have a role in AD.72 Recent investigations have shown a direct relationship between IGF 1/2 and myogenic regulatory factors that allow myogenic stimulus (hypertrophy, atrophy or repair) and IGF release.73,74
Natural NTFs seem to be promising molecules for the treatment of neurological disorders, especially considering their regulatory functions in balancing endogenous cell death and neurorepair processes. They are also responsible for maintaining NVU structure and functions, but despite these roles, until now no large-scale clinical trials of NTFs have been successful.75–77 The pharmacological properties of NTFs render them difficult for clinical application. One of the major obstacles with the therapeutic use of NTFs is the BBB, which usually stops proteins larger than 20 kDa.43,47,78 The problem makes invasive application strategies, such as intracerebroventricular infusion, necessary. Moreover, in the initial clinical trials, several undesirable side effects have been reported (e.g. hyperalgesia, weight loss). The development of alternative neurotrophic therapies employing small molecules that could mimic NTFs or otherwise stimulate selected elements of their signal transduction pathways remains an important goal in neurology.
Experimental Neurotrophic Therapies and Neurotrophic-like Agents Available for Clinical Use
The category of ‘neurotrophic treatment’ is currently considered experimental due to the fact that there is no drug in clinical use that has been registered/approved as ‘a neurotrophic treatment’ per se.


Glatiramer acetate (GA), an immunomodulatory agent approved for the treatment of multiple sclerosis, has been shown to interfere with neurotrophic signalling,79 although no clear mechanism of action has been established for this agent. It consists of randomly synthesised mixture of peptides (6.4 kD average size) derived from four amino acids that are abundant in myelin basic protein: glutamic acid, lysine, alanine and tyrosine. GA is administered peripherally as a subcutaneous injection.
The experimental therapies broadly described as neurorestorative, including cell-based and pharmacological treatments, are currently in different phases of preclinical and clinical development.80,81 These approaches were shown to stimulate angiogenesis, neurogenesis, remyelination, cell migration, suppression of apoptotic-like processes and recovery of functional NVU. The findings at molecular and cellular level translated into improved functional outcomes in various animal models of neurological diseases. They were shown to act through sonic hedgehog (Shh) and neurotrophic signal transduction pathways, which are part of endogenous mechanisms of neuroprotection and neurorepair described previously.82–84
Cerebrolysin is another neuropeptide preparation (referred to below as ‘the neurotrophic factor preparation’ [NTF-prep]) produced by an enzymatic breakdown of purified porcine brain proteins and contains a complex mixture of <10 kDa peptides including fragments of ciliary NTF (CNTF), GDNF and insulin-like growth factors 1 and 2.85 In the experimental models of stroke, TBI and dementia, the NTF-prep acts in a similar way to endogenous NTFs but, favourably, it is able to cross the BBB. Additionally, it was recently established that the NTF-prep amplifies endogenous recovery processes after stroke and TBI as well as counteracts neurodegenerative processes in experimental models of AD.86–90 In the brain injury models, this action was abolished using the selective inhibitor of Shh (cyclopamine), confirming that the NTF-prep presents a unique opportunity for developing novel clinical protocols along with the mentioned earlier group of experimental neurorestorative treatments.90 The NTF-prep displays pleiotropic effects after peripheral intravenous administration, acting at the level of NVU, preventing neuronal degeneration, restoring neuronal cell structure, increasing synaptic density and stimulating neurogenesis and remyelination. These pharmacological properties were shown to promote learning and memory performance and functional recovery as tested in diverse experimental set-ups.91–93 A theoretical advantage of the drug is that its pharmacodynamic action lasts from hours to months. The mechanism of action of NTF-prep is described in recent reviews.94,95 The NTF-prep is approved by National Drug and Food Technology of Argentina for use in AD, vascular dementia, stroke and TBI and is also approved in these indications in several countries in Latin America, Europe and Asia.
To the best knowledge of the authors, the NTF-prep is the only approved drug with neurotrophic mode of action established in extensive experimental investigations. We have overviewed major available clinical data for this neurotrophic agent to assess its potential for prevention/treatment of CIACI. A literature search in PubMed/Medline retrieved 289 articles reporting the use of NTF-prep in different fields, including 32 randomised control trials. The most important are those related to AD and vascular dementias, as well as those investigating therapeutic effects in the brain injuries.

NTF-prep in Alzheimer’s Disease and Related Dementias
An extensive review on the NTF-prep in AD and vascular dementia96,97 identified 12 international, multicentre randomised control studies investigating the therapeutic efficacy of the NTF-prep in these disorders.98–110 These have shown statistically significant and clinically relevant treatment effects of the NTF-prep on cognitive, global and functional domains in mild to moderately severe stages of dementia.
A recent Cochrane review concluded that NTF-prep may have positive effects on cognitive and global function in elderly patients with vascular dementia of mild to moderate severity. It concluded, however, that there is insufficient evidence to recommend NTF-prep as a routine treatment for vascular dementia due to the limited number of included trials, the wide variety of treatment durations and short-term follow-up in most of the trials.111
A new meta-analysis of six randomised controlled trials (RCTs) in AD confirmed favourable benefits–risks relation of the NTF-prep in mild to moderate AD patients treated with a daily dose of 30 ml.103 The analysis showed a significant advantage of the NTF-prep in cognitive and global endpoints versus placebo, with the number needed to treat (NNT) for benefit of 2.9 with respect to the 6-month global clinical change and the calculated NNT for harm of 501 with respect to risk (‘patients with premature discontinuation due to adverse effects [AE]’).
NTF-prep in Brain Injuries
An extensive review identified five randomised controlled studies investigating the therapeutic efficacy of the NTF-prep in stroke.112–116 One of these, the Cerebrolysin Acute Stroke Trial Asia (CASTA) study included 1,070 patients with acute hemispheric ischaemic stroke.114 There was no significant difference in modified Rankin Scale, Barthel Index and the National Institute of Health Stroke Scale (NIHSS) 90 days after stroke onset between patients receiving the NTF-prep or placebo. A post-hoc analysis, however, showed a trend in favour of the NTF-prep in patients with a NIHSS score >12 (cumulative mortality at 90 days was 20. 2 % in the placebo group and 10.5 % in the treatment group; morbidity was lower in the treatment group with an improvement of 4.8 points on the NIHSS versus 1.8 points for placebo). In the most recent RCT, investigating the cognitive effects of the NTF-prep in mild TBI patients, it was found that the acute administration of the NTF-prep resulted in the recovery of cognitive deterioration as assessed at 1 month and 3 months post-injury.117 These results confirmed previous findings118 and suggest independently that the hypothesis of neurotrophic treatment to prevent CIACI deserves dedicated research. Pandharipande et al.2 observed that CIACI presents a similar clinical picture to that observed in moderate TBI patients suggesting involvement of the overlapping pathological mechanisms.
NTF-prep and Safety
Recent comprehensive reviews of NTF-prep in AD analysed safety of the treatment in clinical studies with good scientific quality.94,103 All of the studies demonstrated the safety of the treatment without significant AEs compared with placebo and significant improvements in clinical and cognitive function in the groups treated with NTF-prep. Across several clinical trials, the AEs produced by NTF-prep were generally mild and transient, including dizziness, shaking and feeling hot. Prescribing
information warns of hypersensitivity or allergic reactions in <1/10,000 patients. NTF-prep also appears to be safe when used in combination with recombinant tissue plasminogen activator (rtPA) or cholinesterase inhibitors, such as donepezil or rivastigmine.100,116
Patient Stratification and Sound Clinical Trial Design in CIACI
The growing importance of patients’ stratification in neurology is a direct consequence of a few decades of failure in developing effective therapies for major diseases. Stratification makes sense even in cases where patients’ benefits have been confirmed for a given treatment in dedicated RCTs, and approved. For example, only one in seven ischaemic stroke patients benefits from treatment with thrombolysis. The key factors facilitating stratification of patients’ population are genetic testing and increasing use of a variety of biomarkers. It is believed, for example, that progress in the treatment of AD depends on the development of markers allowing for preventive treatment in the high-risk group of healthy/ mildly affected individuals, long before clinical symptoms of AD can be detected. However, additional demographic, clinical and patient-centred factors can also make major (and sometimes dominant) contributions to development of effective novel clinical protocols.119
In this context, the finding by Pandharipande et al.2 that development of CI can be linked to delirium and intubation, as independent risk factors, justifies the testing of already available and new treatments in this selected group of patients. The pathological features underlying development of CI in this group of patients, past experience in clinical development in neurology as well as the latest developments in understanding the biology of the brain must be taken into account when choosing the most appropriate treatment strategy. The authors believe that the available research and clinical data point to the neurotrophic approach as a compelling therapeutic option for prevention/treatment of CIACI.
Conclusion
CIACI is an example of a pathophysiological mechanism in which secondary injury generates neurological lesions in a previously healthy brain. This damage can be seen in both pathological2,5–7,12,120 and nuclear magnetic resonance (NMR) imaging studies.14–17 Cerebral atrophy, whitematter hyperintensity and leukoaraiosis are as common in vascular and mixed dementia as they are in CIACI, whereas ventricular dilatation and atrophy, especially of the hippocampus, are both identified in CIACI and AD. This relationship has led some to suggest that increased rates of AD are partly driven by the effects of critical illness and its treatment.121 These findings should be analysed not only in terms of mechanisms of cell damage, sources of excitotoxicity, inflammation, necrosis, apoptosis, but also in terms of endogenous brain repair processes. The context of NVU should be always observed for better understanding the condition and, consequently, for devising new treatments.49,50
Longer durations of delirium are associated with worse overall cognition and poorer executive function.2 This finding indicates significant progress in the critical care patients’ stratification for the improved clinical protocols. The hypothesis of delirium generation involves more than one DM, and could support the use of pleiotropic therapeutic agents (see Figure 6) with neuroprotective and neurorestorative properties.94,122
Current prevention strategies and treatments, including tests of awakening and physical and cognitive rehabilitation, are therapeutic approaches that promote neuroprotection and neurorehabilitation. The endogenous repair processes can be stimulated using arousal methods, cognitive stimulation and neuromuscular activity that activate the release of NTFs.66–69,72–74,123,124 Neurotrophic agents may be administered when a patient arrives at the ICU and during the critical period; their pharmacological and physiological properties make them suitable for use in the prevention and treatment of CIACI. The time frame of brain DMs in critically ill patients and the therapeutic windows of opportunity that could be exploited by such a treatment are summarised in Figure 7. The increased understanding and recognition of CIACI and advances in critical care treatment support the initiation of appropriate randomised prospective trials. Such investigations are much needed since there is currently no proven treatment for this disease, which is a substantial and increasing public health problem.