
The baroreflex is one of the most important neural mechanisms involved in blood pressure regulation. Baroreceptors in the carotids, cardiac chambers and the aortic arch are activated by changes in blood pressure. The information from baroreceptors relays in the nucleus tractus solitarius and the ventrolateral medulla and is further processed in regions including the insula, medial prefrontal cortex, cingulate cortex, amygdala, hypothalamus and cerebellum.1 By adjusting the heart rate (vagal) and peripheral vascular tone (sympathetic), the baroreflex compensates spontaneous fluctuations in blood pressure.
Impairment of blood pressure regulation resulting in blood pressure elevation or instability is a frequently observed phenomenon in acute stroke. In general, underlying pathophysiological mechanisms are unknown; however, there is increasing evidence for central autonomic system involvement, particularly sympathetic system overdrive activation and impairment of the baroreceptor reflex arch. Baroreflex failure may be caused by impairment of baroreceptors, afferent baroreceptive nerves or central processing. Autonomic disorders, carotid endarterectomy, bilateral atherosclerosis of carotid arteries, brainstem stroke and acute hemispheric stroke are all recognised causes of baroreflex impairment.2–6 Of clinical importance, impaired cardiac baroreceptor reflex sensitivity (BRS) has been shown to independently predict mortality after myocardial infarction and was associated with poor prognosis of chronic heart failure or multiple organ dysfunction syndrome.7–9 Similarly, baroreflex impairment has been independently related to less favourable long-term outcome after acute ischaemic stroke10 or after intracerebral haemorrhage.11 In a recent study of patients suffering spontaneous intracerebral haemorrhage, we demonstrated a significant and independent association between impaired BRS, beat-to-beat blood pressure variability, clinical blood pressure variability and short-term outcome. Traditionally, baroreflex impairment has been linked with clinical manifestations as hypertensive crisis, rapid blood pressure fluctuations or volatile hypertension. These conditions are known to occur also in the acute phase of haemorrhagic or ischaemic stroke and appear to negatively influence te outcome.12,13 Dawson et al. reported that poor outcome at 30 days after ischaemic stroke was dependent on blood pressure variability obtained within the first 72 hours of ictus.14 Stead et al. showed that wide fluctuations of blood pressure in the first three hours of acute ischaemic stroke appeared to be associated with increased risk of death at 90 days in a cohort of 71 patients.13 As autoregulation seems to be impaired in both acute ischaemic and haemorrhagic stroke, fluctuations in blood pressure may significantly alter cerebral perfusion, leading to secondary brain injury.15–17 Moreover, associations between hypertensive crisis at admission and haemorrhage enlargement or oedema formation have been described.18,19
As baroreflex seems to be blunted by both carotid atherosclerosis and lesions affecting central processing, controversy exists in terms of the aetiology of stroke-related baroreflex changes.20 However, in our previous study, baroreflex impairment in acute stroke could be seen independently of carotid atherosclerosis. We have demonstrated decreased baroreflex function in stroke patients compared with controls matched for atherosclerosis and other possible confounding factors, which we therefore attribute to lesions affecting central processing. Moreover, the central role of the insula in baroreflex regulation could be demonstrated. Both insulae seem to participate in baroreceptor information processing in a complex manner, with the left insula being more dominant in baroreflex control, presumably through parasympathetic outflow modulation.21
How baroreflex dysfunction influences outcome in acute stroke has not yet been clearly established. Patients with shifted autonomic balance seemed to be at increased risk of cardiac complications and have significantly higher cardiovascular morbidity and mortality.8,10,22 Suggested pathophysiological mechanisms include raised arrhythmogenic potential, increased platelet aggregability, coronary vasoconstriction and impaired ventricular remodelling, all of which are thought to be associated with increased sympathetic activity. Moreover, autonomic impairment possibly plays an important role in secondary brain injury after stroke. Decreased baroreflex sensitivity and impaired blood pressure regulation are consistent with increased sympathetic activation. A shift to sympathetic predominance has been previously shown to be associated with pro-inflammatory cytokine production, hyperglycaemia and increased blood–brain barrier permeability.23–25 In turn, these mechanisms have been proposed to be involved in brain oedema formation and secondary brain injury.26–28 Arterial baroreflex function has been shown to be an important determinant of acute cerebral ischaemia in rats with middle cerebral artery occlusion. Baroreflex dysfunction significantly increased the levels of the pro-inflammatory factors interleukin (IL)-1 and IL-6 and the infarct volume.29 Therefore, the question of therapeutic modulation of autonomic baroreflex dysfunction with the aim of influencing outcome of stroke seems to become relevant. Baroreflex can be positively influenced by pharmacological treatment, especially with beta-blockers.30,31 Interestingly, a recent study by Loawattana et al. including 111 ischaemic stroke patients showed that beta-blocker use was independently associated with less severe stroke on presentation and that sympatholytic effects may have cerebroprotective properties.32 In an animal model, beta-blockers given before experimental ischaemia reduced infarct volume by 40%.33 Analogously, beta-blockers reduced histologic brain oedema in a model of traumatic brain injury.34 Positive effects of beta-blockade on outcome have also been reported in traumatic brain injury patients.35,36
In the study by Liu et al., ketanserin in doses of 3mg/kg per day decreased blood pressure and enhanced BRS, whereas 0.3mg/kg per day only enhanced the BRS. Fatal stroke incidences were markedly reduced by treatment with both doses (p<0.0001 versus control group).37 Clonidine, moxonidine, folic acid and mecobalamin may improve baroreflex sensitivity as well. Central mechanism of action is supposed in the effects of clonidine and moxonidine, versus peripheral mechanism in mecobalamin.38,39 New devices stimulating baroreceptors are emerging in the treatment of chronic refractory hypertension. Mediated through the central sympathoinhibitory effect by stimulating the carotid baroreceptors electrically, these devices ameliorate baroreflex sensitivity and reduce hypertension. Ongoing trials are finding significant and sustained reductions in blood pressure, a good safety profile and tolerable side effects;40 however, the evidence is still scarce.
Whether baroreflex modulation may change the course of acute stroke remains an important but unresolved question. The implications of autonomic dysfunction in acute stroke, including baroreflex impairment, are poorly understood – in particular how autonomic derangement effects outcome in stroke. However, previous studies uniformly confirm the association between autonomic impairment and worse short- or long-term outcome after ischaemic or haemorrhagic stroke. Unfortunately, the proposed underlying mechanisms are still speculative. More studies focusing on this topic are urgently required. Nevertheless, it definitely seems that baroreflex should become a novel research and potentially also therapeutic target in acute stroke. ■
Modulation of Baroreceptor Reflex Sensitivity May Represent a New Therapeutic Target in Acute Stroke
Abstract
Overview
Acute stroke has been associated with several manifestations of autonomic dysfunction including cardiovascular, sudomotor, thermoregulatory, gastrointestinal and urogenital symptoms. In particular, cardiovascular autonomic impairment including blunted baroreflex has been repeatedly shown to be of prognostic importance in acute stroke. Pathophysiological mechanisms of baroreflex changes in acute stroke include lesions of the central autonomic processing with consequent sympathetic system overactivation and impairment of baroreflex functioning. Previous studies have shown that patients with shifted autonomic balance are more prone to develop cardiac complications and have increased cardiovascular morbidity and mortality. Moreover, autonomic dysregulation may play an important role in secondary brain injury after stroke. Therefore, modifying autonomic functions may have important therapeutic implications in acute stroke. In this article, the role of baroreflex impairment in acute stroke and its possible therapeutic relevance is discussed.
Keywords
Baroreflex, autonomic, ischaemic stroke, intracerebral haemorrhage, outcome, therapy
Article
References
- Henderson LA, Richard CA, Macey PM, et al., J Appl Physiol, 2004;96:693–703.
- Nouraei SA, Al-Rawi PG, Sigaudo-Roussel D, et al., J Vasc Surg, 2005;41:631–7.
- Schrezenmaier C, Singer W, Swift NM, et al., Arch Neurol, 2007;64:381–6.
- Phillips AM, Jardine DL, Parkin PJ, et al., Stroke, 2000;31:1997–2001.
- Robinson TG, James M, Youde J, et al., Stroke, 1997;28:1671–6.
- Nasr N, Pavy-Le Traon A, Larrue V, Stroke, 2005; 36:1891–5.
- La Rovere MT, Bigger JT Jr, Marcus FI, et al., Lancet, 1998;351:478–84.
- Mortara A, La Rovere MT, Pinna GD, et al., Circulation, 1997;96:3450–58.
- Schmidt H, Muller-Werdan U, Hoffmann T, et al., Crit Care Med, 2005;33:1994–2002.
- Robinson TG, Dawson SL, Eames PJ, et al., Stroke, 2003;34:705–12.
- Sykora M, Diedler J, Rupp A, et al., Crit Care Med, 2008;36:3074–9.
- Willmot M, Leonardi-Bee J, Bath PM, Hypertension, 2004;43:18–24.
- Stead LG, Gilmore RM, Vedula KC, et al., Neurology, 2006;66:1878–81.
- Dawson SL, Manktelow BN, Robinson TG, et al., Stroke, 2000;31:463–8.
- Immink RV, van Montfrans GA, Stam J, et al., Stroke, 2005;36:2595–2600.
- Kuwata N, Kuroda K, Funayama M, et al., Neurosurg Rev, 1995;18:237–45.
- Diedler J, Sykora M, Rupp A, et al., Stroke, 2009;40(3):815–19.
- Vemmos KN, Tsivgoulis G, Spengos K, et al., J Hypertens, 2003;21:2167–73.
- Kazui S, Minematsu K, Yamamoto H, et al., Stroke, 1997;28:2370–75.
- Oppenheimer S, Stroke, 2003;34: 705–12.
- Sykora M, Diedler J, Rupp A, et al., Stroke, 2009;40(3):737–42.
- La Rovere MT, Specchia G, Mortara A, Schwartz PJ, Circulation, 1988;78:816–24.
- van der Poll T, Lowry SF, Infect Immun, 1997;65:2378–81.
- Watanabe M, Tomiyama-Miyaji C, Kainuma E, et al., Immunol Lett, 2008;115:43–9.
- Raichle ME, Hartman BK, Eichling JO, Sharpe LG, Proc Natl Acad Sci U S A, 1975;72: 3726–30.
- Castillo J, Davalos A, Alvarez-Sabin J, et al., Neurology, 2002;58:624–9.
- Song EC, Chu K, Jeong SW, et al., Stroke, 2003;34:2215–20.
- Xi G, Keep RF, Hoff JT, Lancet Neurol, 2006;5:53–63.
- Liu AJ, Ling G, Wu J, et al., Life Sci, 2008;83: 388–93.
- Elghozi JL, Julien C, Fundam Clin Pharmacol, 2007;21:337–47.
- Mortara A, La Rovere MT, Pinna GD, et al., J Am Coll Cardiol, 2000;36:1612–18.
- Laowattana S, Oppenheimer SM, Neurology, 2007;68:509–14.
- Savitz SI, Erhardt JA, Anthony JV, et al., J Cereb Blood Flow Metab, 2000;20:1197–1204.
- Liu MY, J Formos Med Assoc, 1995;94:386–90.
- Inaba K, Teixeira PG, David JS, et al., J Am Coll Surg, 2008;206:432–8.
- Cotton BA, Snodgrass KB, Fleming SB, et al., J Trauma, 2007;62:26–33, discussion 33–25.
- Liu AJ, Ma XJ, Shen FM, et al., Stroke, 2007;38:1916–23.
- Ma XJ, Shen FM, Liu AJ, et al., Acta Pharmacol Sin, 2007;28:1550–58.
- Turcani M, J Cardiovasc Pharmacol, 2008;52:524–35.
- Uppuluri SC, Storozynsky E, Bisognano JD, Curr Hypertens Rep, 2009;11:69–75.
Article Information
Disclosure
The authors have no conflicts of interest to declare.
Correspondence
Marek Sykora, Department of Neurology, University of Heidelberg, Im Neuenheimer Feld 400, 69120 Heidelberg, Germany. E: marek.sykora@med.uni-heidelberg.de
Received
2009-02-22T00:00:00
Further Resources

Trending Topic
Both intracerebral hemorrhage and ischemic stroke continue to be the leading causes of disability and the second leading causes of death worldwide.1,2 The burden is largest in low- and middle-income countries, which have seen rapid recent population growth. Large vessel occlusion (LVO – internal carotids, vertebrals and the proximal branches of the circle of Willis) accounts for 20% of all acute ischemic strokes (AISs) […]
Both intracerebral hemorrhage and ischemic stroke continue to be the leading causes of disability and the second leading causes of death worldwide.1,2 The burden is largest in low- and middle-income countries, which have seen rapid recent population growth. Large vessel ...

It is my pleasure to introduce the 2025 of touchREVIEWS in Neurology, which brings together an exceptional collection of reviews, original research and congress highlights that reflect the continued evolution of neurological science and clinical innovation. This issue opens with Jelle ...
Acute ischaemic stroke management Recent randomized controlled trials (RCTs) comparing bridging intravenous thrombolysis (IVT) with tissue plasminogen activator (tPA) to direct endovascular treatment (EVT) for large-vessel occlusion (LVO) strokes within 4.5 hours of onset have failed to show inferiority of direct ...

The prevalence of unruptured intracranial aneurysms (IAs) is approximately 3% of the population, with incidence on the rise due to the increased utilization of neuro-imaging for diverse objectives.1,2 The average risk of rupture for unruptured IA is estimated to vary from 0.3% ...
What is the Stroke Action Plan for Europe? Stroke is one of the most enormous burdens to healthcare services.1 Despite our combined efforts, it affects more than one million people annually in Europe. Although we have abundant knowledge regarding stroke ...

Introduction The term 'stroke' is used to describe an adverse clinical state involving interference of blood circulation to the brain due to obstruction or rupture of blood vessels.1 Stroke was previously categorized into a cardiovascular disorder until the release of ...
In clinical practice, the definition of penumbra is largely based on neuroimaging and indicates potentially salvageable tissue. Perfusion imaging has been useful in identifying this tissue when obeying ‘the mismatch concept’. The mismatch concept is a surrogate marker for salvageable ...

Ischaemic stroke may occur due to several potential mechanisms. Mechanisms are classically described as large artery atherosclerosis, small-vessel occlusion, cardioembolism, stroke of other determined aetiology (i.e. vasculitis, genetic disorder, etc.) and stroke of undetermined aetiology. Historically, the definition of ...
Cerebral amyloid angiopathy (CAA), also known as congophilic angiopathy, is a recognized cause of lobar intracerebral haemorrhage in patients above the age of 50 years.1 It is known to be due to deposition of a variant of amyloid (beta-amyloid plaque) in ...
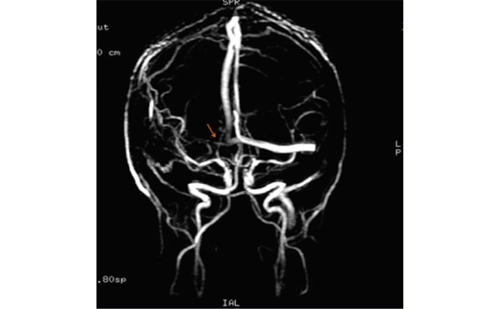
Cerebral venous thrombosis (CVT) is a rare condition.1–3 While thrombophilias, contraceptive pill, hormonal replacement therapy, and neoplasms are well-established predisposing factors, steroid use is a less-common possible risk factor, particularly with regard to anabolic androgenic steroids (AAS).1–4 We intend to ...
Globally, a high proportion (around 25%) of transient ischemic attacks (TIAs) and ischemic strokes are cryptogenic.1,2 The Trial of ORG 10172 in Acute Stroke Treatment (TOAST) classification defines a cryptogenic stroke as a brain infarction that is not caused by definite cardioembolism, ...
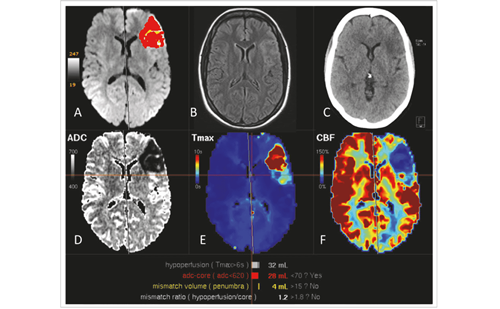
In 2018, two randomised controlled trials, DAWN (the Clinical Mismatch in the Triage of Wake up and Late Presenting Strokes Undergoing Neurointervention with Trevo Thrombectomy Procedure) and DEFUSE 3 (Endovascular Therapy Following Imaging Evaluation for Ischaemic Stroke 3), implemented computed tomography (CT) and ...
Log into your Touch Account
Keep track of your clinical interests and newsletter subscriptions.
Sign up with an Email
Or use a .
Register now for FREE access
Already registered? Login below.
