Every year in the US there are approximately 30,000 reported cases of aneurysmal subarachnoid hemorrhage (SAH), accounting for 5% of all strokes.1–3 Mortality has been documented in the range of 20–40% and over 40% experience long-term deficits.2,4,5 Although SAH is less common than diseases such as ischemic stroke and heart attack, it affects a younger population and contributes to a disproportionately high ratio of long-term disability after stroke. SAH is responsible for 27% of all strokerelated potential life-years lost before 65 years of age.6 Long-term neurological injuries in SAH arise from the acute injuries due to sudden aneurysm rupture and, in a significant fraction of SAH patients, secondary neurological injuries due to delayed cerebral vasospasm. Past literature has often referred to this secondary neurological insult as delayed cerebral ischemia (DCI). Recent studies on vasospasm have demonstrated that DCI, defined differently in various studies, likely includes both reversible states of cerebral ischemia and completed strokes. As a result, current literature has moved away from using the term DCI in studying SAH and vasospasm.
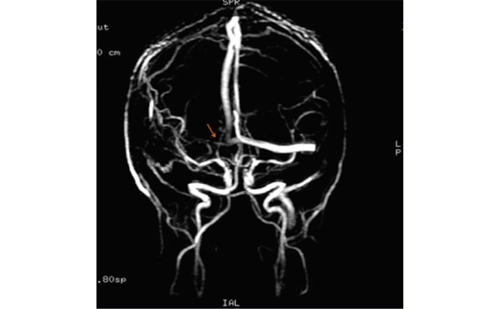
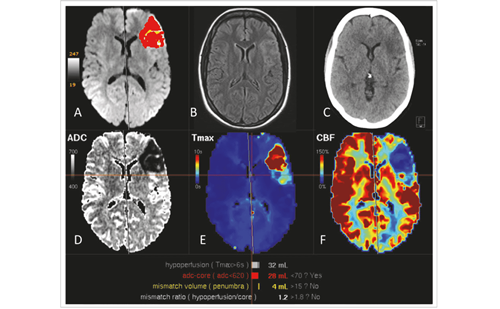
The definition of vasospasm is variable in the literature. Guidelines from the American Heart Association (AHA) in 2009 defined cerebral vasospasm as “the delayed narrowing of large-capacitance arteries at the base of the brain.”3 Onset of vasospasm has been reported to occur somewhere between days four and 15 post-SAH.5,7 There are various diagnostic criteria for cerebral vasospasm following SAH. Symptomatic or clinical vasospasm is often defined as neurological deterioration of greater than 2 points in the Glasgow Coma Scale without apparent etiology other than cerebral ischemia.8 In these cases, delayed symptomatic vasospasm has also been referred to as DCI and is reported to occur in 15–40% of patients after aneurysmal SAH.3,9 Vasospasm can be defined by transcranial Doppler ultrasonography (TCD) criteria; most centers use a Lindegaard ratio of >3–4.5 to indicate mild, 4.5–6 to indicate moderate, and >6 to indicate severe vasospasm in the anterior circulation. Vasospasm as recorded by TCD is reported to occur in 46–70% of patients after SAH, and is defined by either peak or mean flow velocity.4,7 Four-vessel diagnostic cerebral angiography is currently the gold standard for radiographic detection of vasospasm, and angiographic vasospasm has been documented to occur in 30–70% of patients after aneurysmal SAH (see Figures 1 and 2).3 Other radiographic diagnostic modalities such as computed tomography angiography (CTA) and perfusion magnetic resonance imaging (MRI) with angiography are also used to detect vasospasm.
Currently, few therapies can be used to prevent vasospasm. Oral nimodipine, a calcium channel antagonist, appears to improve overall outcome in SAH patients but does not seem to influence the rate of symptomatic vasospasm when used in prospective trials.10–12 Intravenous nicardipine has been shown to decrease the rate of symptomatic and angiographic vasospasm but did not improve patient outcome when used in randomized studies.13,14 Once vasospasm occurs, current medical therapy for treatment of the vasospasm is the so-called ‘triple-H therapy,’ which includes induced hypervolemia, hypertension, and hemodilution.15–17 This therapy has unproven efficacy, requires intensive care unit (ICU) treatment, and can cause complications such as heart failure and myocardial injuries. Severe vasospasm refractory to medical treatment may be treated with intra-arterial injection of calcium channel blockers or balloon angioplasty.18,19 However, these are highly invasive procedures with associated risk for stroke, cerebral artery dissection, or rupture, and even death.
Statins, which are hydroxymethylglutaryl coenzyme A inhibitors, have been in the news lately for their potential effects in preventing cardiovascular and neurological diseases such as myocardial infarction and stroke.20 Statins are typically prescribed to lower cholesterol because of their ability to inhibit cholesterol synthesis, and their safety and efficacy have been investigated when used to prevent coronary heart disease.4 Over the years, scientists have discovered many other actions of statins. The pleiotropic effects of statin drugs appear to target many pathogenic mechanisms implicated in the causation of cerebral vasospasm.21 The beneficial effects of statins in cerebral vasospasm have been shown in animal models, and there are now several human studies.
Mechanism of Vasospasm
Vasospasm onset has been associated with a range of symptoms, from mild to severe, including focal deficits such as visual problems, hemiparesis, hemiplegia, difficulty speaking, and ambiguous symptoms such as malaise and confusion.7 Many different pathophysiological mechanisms for vasospasm have been proposed, including endothelial dysfunction, nitric oxide (NO) deficiency, increased oxyhemoglobin and bilirubin oxidation product (BOXes) in the subarachnoid space, oxidative stress, endothelin-1 elevation, inflammation, microthrombi formation, and vascular smooth-muscle dysfunction.9,22 After an aneurysm ruptures into the subarachnoid space, blood and hemoglobin breakdown products are suddenly present where they are not typically found. Current theories postulate that the presence of these compounds leads to an imbalance of cerebral vasoconstrictors and vasodilators, causing vasospasm. One of the well-studied pathways is the interaction between oxyhemoglobin and NO. Oxyhemoglobin is a strong NO scavenger system and binds to NO with high affinity in the subarachnoid space, leading to a deficiency in cerebral vasodilation.23 Oxyhemoglobin in the subarachnoid space is metabolized into hemoglobin breakdown products including BOXes, which are potent vasoconstrictors.9,24 Furthermore, oxyhemoglobin also causes the release of superoxide free radicals, which can inhibit NO activity and cause oxidative stress.9
Another effect of the aneurysm rupture is endothelial dysfunction and an increase in endothelin-1 (ET-1) release from the vascular endothelium. ET-1 is one of the most potent vasoconstrictors in the central nervous system (CNS), and excess ET-1 likely contributes to the development of vasospasm.23,25 Furthermore, blood and hemoglobin breakdown products cause an inflammatory response that sets off prostaglandin imbalance and cytokine release, which contributes to the dysregulation of vascular smooth-muscle tone.9,26 Despite characterization of all these pathways, the overall mechanism of vasospasm remains unresolved and is an active area of research. Recent studies have implicated other pathogenic mechanisms in vasospasm such as platelet activation, neuronal apoptosis, and microthrombi formation, to name a few.9,25,27 Furthermore, the inconsistent correlation between angiographic vasospasm severity and clinical neurological deficit raise the question that mechanisms other than visible large-caliber cerebral arterial constriction may be responsible for secondary neurological injuries and long-term disability after SAH.
How Can Statins Prevent Vasospasm?
Research has demonstrated that statins have many actions in addition to lowering cholesterol. Cardiologists discovered that high-dose statin actually lowers mortality after acute myocardial infarction, stabilizes atherosclerotic plaques, and remodels the left ventricle.21 Statins have pleiotropic effects, many of which appear to target pathogenic mechanisms in vasospasm.21 In the central nervous system, statins have been shown to be anti-inflammatory, reduce oxidative stress, improve endothelial function, enhance fibrinolysis, and improve smooth-muscle-cell function.21,22,28,29 Statins have been shown to increase release of NO by upregulation of endothelial NO synthase and promoting vasodilation.22 Some studies have also suggested that statins can attenuate leukocyte migration to the site of endothelial damage.30 All of these effects target the underlying causes of vasospasm and support the hypothesis and hope that statins may be effective in the prevention and amelioration of vasospasm in humans (see Table 1).
Animal Studies
Animal studies have shown that statins have the potential to reduce vasospasm, DCI, and vasospasm-related infarcts. McGirt et al. showed that mice pre-treated with statins have decreased incidence of vasospasm and better neurological outcome.31 Mice were administered subcutaneous simvastatin or placebo for 14 days, then underwent right anterior cerebral artery perforation or sham surgery. Results showed decreased vasospasm in simvastatin-treated mice versus controls, as shown by a significantly larger right middle cerebral artery (MCA) diameter. Pre-treated mice also had increased cerebral endothelial NO synthase protein and experienced better neurological outcome compared with non-treated mice. This study prompted further investigation because it provided the groundwork by which statins may influence cerebral NO synthesis and therefore play a role in reducing vasospasm.
Although their first study proved statin pre-treatment could decrease vasospasm after SAH in mice, pre-treatment in the human model is not feasible as aneurysmal SAH is difficult to predict. McGirt et al. followed up with another study to investigate statin treatment after SAH in rabbits in order to better model the human disease.30 Researchers injected blood into the cisterna magna of rabbits in order to model a posterior circulation aneurysmal SAH, followed by subcutaneous simvastatin or placebo administration. The results showed that vasospasm, as measured by basilar artery diameter, was decreased in rabbits treated with simvastatin versus control. The data also revealed a significant decrease of inflammation in the cerebral blood vessels.
Human Data
Retrospective Human Data
The last several years have provided clinical data regarding the use of statins in humans to improve outcome after aneurysmal SAH. One of the first studies, conducted by Parra et al., investigated the effect of prior statin use on outcome after SAH.4 This retrospective matched case– control study included 20 SAH patients on statins prior to their aneurysm rupture and onset of SAH matched 1:2 to a control cohort of 40 SAH patients who were not previously on statin therapy. The primary outcome for this study was functional outcome at 14 days post-SAH as measured by Barthel Index and Modified Lawton Physical Self-Maintenance Exam scale scores. Secondary outcomes examined in this study were 14-day mortality, Modified Rankin Scale (mRS), DCI, infarction, or elevated mean velocity on TCD. The statin cohort had significantly better functional outcomes, decreased DCI and other cerebral infarction, and lower mean velocity measured by TCD, indicating a lower incidence of vasospasm. However, the study also showed that mortality and overall outcome (measured by mRS scores) were similar between the groups despite improvements in surrogate markers of vasospasm and cerebral ischemia.
Another study, performed by Singhal et al., examined the effects of various pre-admission medication exposures on the incidence of vasospasm.32 Researchers identified 514 patients who were admitted with aneurysmal SAH between 1995 and 2003. Using logistic regression, the authors found that statin use prior to hospitalization was possibly associated with increased risk for developing vasospasm (p=0.05). On review of their data, the authors discovered that all patients who presented to the hospital on statin had their statin medication stopped after admission for SAH. They postulate that it is the abrupt cessation of statin that may be associated with increased risk for vasospasm, and cautioned against this practice. However, another retrospective study conducted by Moskowitz et al. reported a different result:33 after reviewing aneurysmal SAH patient files from 1997 to 2004, researchers concluded that prior statin use decreased the incidence of angiographic vasospasm post-SAH. They also concluded that discontinuation of statin did not increase incidence of vasospasm, contrary to the Singhal et al. study.
Lastly, Kramer et al. explored empirically the results of a protocol change in May 2006 at the University of Virginia Neuroscience ICU where patients admitted with SAH were administered simvastatin 80mg/day for two weeks.5 They conducted a retrospective cohort study of SAH patients admitted between November 2004 and February 2007, and compared incidence of vasospasm before and after this new protocol. After a review of the charts, 150 patients were included in the analysis, with 71 patients who had received statin. Primary outcomes included radiographic (by CTA) and symptomatic vasospasm, and delayed cerebral infarction. The results revealed that there was no significant difference in improvement seen in the statin-treated versus non-treated group.
Prospective Human Data
There are now several small pilot prospective randomized studies of statin use for the prevention of vasospasm after SAH. Tseng et al. randomized 80 aneurysmal SAH patients in England into a treatment group that received oral pravastatin dosed at 40mg/day or a control group that received placebo.34 Patients began treatment with pravastatin or placebo within 72 hours of stroke onset and were treated for 14 days. The treatment group showed a significant decrease in vasospasm (defined by TCD criteria) of 32% compared with the control group, and also a 42% decrease of severe vasospasm. The results also showed an 83% decrease in DCI. Furthermore, there were no reported risks associated with pravastatin treatment and therefore the study garnered more confidence in the safety of statin use to prevent vasospasm. A follow-up study was conducted by Tseng et al. to compare the outcomes of those who received pravastatin versus placebo at six months post-SAH.35 Researchers found that pravastatin reduced unfavorable neurological outcome by 73% as measured by Short-Form 36 (SF-36) (a measure of mental and physical health) and mRS.
The first US study was conducted by Lynch et al. from Duke University. In this study, SAH patients were randomized to receive simvastatin 80mg/day or placebo for 14 days.36 The primary end-point was defined as vasospasm (by TCD criteria) with clinical manifestation of DCI. The data revealed decreased mean MCA velocity as measured by TCD as well as decreased incidence of vasospasm in the treatment group. The authors also explored plasma von Willebrand factor and serum protein S100β levels as potential markers of endothelial dysfunction and neuronal injury, respectively. Both markers were lower in the treatment group compared with the control group, suggesting that simvastatin treatment reduced the degree of endothelial dysfunction and brain injury in patients post-aneurysmal SAH.
Another pilot double-blind, placebo-controlled study was conducted by Chou et al. to investigate the safety and efficacy of simvastatin with a dosage of 80mg/day.8 Thirty-nine patients not previously treated with statins were randomized into treatment and control groups within 96 hours of aneurysmal SAH. This study looked at a more select SAH population with Fisher grade 3 or higher. While simvastatin at 80mg/day appears safe to use in this patient population, Chou et al. did not find significant differences in the incidence of vasospasm by clinical, TCD, or angiographic criteria. There was no difference in the incidence of stroke or functional outcomes between groups. Although all prospective human trials thus far have had small sample sizes, Sillberg et al. completed a meta-analysis based on available randomized study data.1 Data analysis revealed that the use of statins in patients post-SAH decreased the incidence of vasospasm and DCI, as well as mortality.
At this point it is important to keep in mind the variations in study design, as this could explain the variety of results thus far. One source of variation is the definition and diagnosis of vasospasm between the studies. The small sample sizes have also influenced the ability to reach statistical power in some cases. Further complicating the data is the variation in time to starting the study drug, and also the diverse patient populations and different treatment protocols of vasospasm in SAH. Of note, all of the studies to date have used maximal US Food and Drug Administration (FDA)-approved statin dosage, which is approximately 10% of the dosage used in animal studies by weight-based calculation. The optimal dosage of statin to use in trials studying its effect against vasospasm is not known at this time.
Conclusion
Statins have been shown to be safe for use in patients post-aneurysmal SAH. Multiple animal studies and two pilot prospective randomized human studies have suggested potential beneficial effects of statin for prevention and treatment of vasospasm following SAH. Other studies of similar sample size did not demonstrate drug efficacy. To date, statin use for prevention of delayed vasospasm and secondary neurological injuries following SAH remains a therapeutic option with potential efficacy, but with mixed results from small pilot studies. The role of statin use in vasospasm treatment is one of the most important unanswered questions in neurocritical care and is ready for a collaborative multicenter phase III randomized study. Studies on vasospasm and neurological outcome to date have not shown consistent results, partly due to a wide variety of definitions used for vasospasm and neurological outcomes across different research groups and institutions. It is important that the community come to a consensus on the definitions of vasospasm and agree on measures of clinical outcome. Furthermore, the varying results from human studies to date may be due to small sample size, but it is also possible that statin may have a short treatment window and therapeutic dose window in its effect on vasospasm. Important questions to be answered in the future by larger human trials include the best timing for statin initiation, the optimal dose of statin, and the mechanistic function of statins on vasospasm pathogenesis in SAH. ■