
Between 60% and 80% of cases with dementia worldwide are caused by Alzheimer’s disease (AD), making it the most prevalent type of dementia.1 As of 2025, approximately 60 million people worldwide are affected by dementia, and by 2050, projections suggest a rise to nearly 210 million.2 Age is the strongest risk factor, with AD affecting nearly half of those aged >85 years.3 In both the USA and Europe, the prevalence of AD increases with age, from 0.85% among individuals aged 65–69 years to 44.35% in those aged over 95 years.4
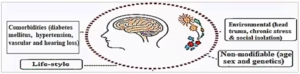
In addition to ageing, Figure 1 shows several other factors that contribute to AD risk, including female sex, genetic predisposition, low educational attainment, head injuries and lifestyle-related behaviours such as a sedentary lifestyle, smoking, alcohol use and obesity. These risk factors synergistically interact with ageing to promote the development of AD.5
Figure 1: Risk factors of Alzheimer’s disease
Despite years of research, the treatment of AD remains limited. This highlights the need for novel approaches that target the underlying pathophysiology. The biggest risk factor for AD is ageing, and targeting ageing processes may be more successful than amyloid-beta (Aβ)/phosphorylated tau (p-tau) strategies in altering the course of AD. This article critically evaluates the biological pathways through which ageing contributes to AD pathogenesis and examines innovative lifestyle and pharmacological interventions that target these processes.
Methods
This narrative review employed a structured, two-phase screening process. An initial screen of 250 titles and abstracts led to the exclusion of 150 records. A subsequent full-text review of the remaining 100 articles identified 30 duplicates, resulting in 79 studies for inclusion. A structured literature search was undertaken to identify these publications. Electronic databases (MEDLINE via PubMed, Embase, Cochrane Central Register of Controlled Trials, and Google Scholar) were searched for articles published from January 2015 through September 2025.The search strategy utilized a comprehensive set of keywords encompassing the core concepts of the review: the biology of ageing (e.g. ‘biological aging,’ ‘geroscience,’ ‘microglial senescence’), Alzheimer’s disease pathology (e.g. ‘amyloid-beta,’ ‘tau,’ ‘neuroinflammation’), and potential interventions (e.g. ‘senolytics,’ ‘epigenetics,’ ‘lifestyle interventions’).
Inclusion criteria
- Study types: longitudinal cohort studies, randomized controlled trials, meta-analyses, systematic reviews and preclinical studies in animal models. Only published articles were included.
- Population: studies focusing on ageing mechanisms and their contribution to AD pathogenesis.
- Intervention/exposure: research on interventions targeting hallmarks of ageing (e.g. cellular senescence, epigenetic alterations, loss of proteostasis) in the context of AD.
- Outcome: measurable outcomes related to AD pathology (Aβ, p-tau), cognitive function or biomarkers of ageing.
Exclusion criteria
- Studies published before 2015 or not available in English.
- Articles not peer-reviewed (e.g. editorials, commentaries without original data, conference abstracts).
- Studies focusing on non-age-related forms of dementia (e.g. familial AD mutations without an ageing context, vascular dementia alone).
The study selection process involved an initial screening of titles and abstracts, followed by full-text review of potentially relevant articles. Data from the included studies were extracted and synthesized to provide a critical overview of the field. As a narrative review, this work may be subject to selection and reporting bias. I attempted to mitigate this by including multiple databases and emphasizing both supportive and negative findings.
Alzheimer’s disease pathology: Possible role of ageing
Ageing induces systemic biological changes that disrupt homeostasis at the molecular, cellular and tissue levels. These alterations include mitochondrial dysfunction, impaired proteostasis, genomic instability, oxidative stress, inflammation and reduced regenerative capacity. In the brain, ageing disrupts proteostasis, synaptic function, immune surveillance, metabolism and the blood–brain barrier’s (BBB) integrity.3 The formation and clearance of Aβ and p-tau, neurotransmitter synthesis and release, microglial and astrocyte responses, neuronal excitability and plasticity, gut microbiota functions and autophagy are all rapidly disrupted by these age-related changes – including oxidative stress, mitochondrial dysfunction and BBB breakdown – that disrupt protein clearance, synaptic function and immune regulation, ultimately driving AD progression (Figure 2).4
Figure 2: Pathology of Alzheimer’s disease
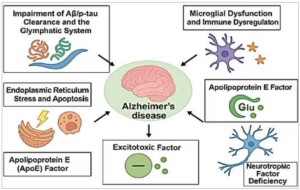
Aβ/p-tau = amyloid-beta/phosphorylated tau; APOE = apolipoprotein E.
Impairment of the amyloid-beta/phosphorylated tau clearance and the glymphatic system
Aβ is an acquired biomarker that can be used to predict disease because it appears long before the clinical signs of AD.6 At normal levels, Aβ has numerous vital functions, including neuroprotection, memory improvement and neuronal growth and repair. The accumulation of Aβ plaques is due to an imbalance between production and clearance. Its accumulation activates immune responses, causing neuronal destruction and memory impairment.6 p-tau interacts with Aβ, exacerbating neurodegeneration through processes such as hyperphosphorylation.5 Sleep may actively remove poisons and metabolites such as Aβ and p-tau from the brain, according to certain theories. However, according to Miao et al., anaesthesia, sleep and awake do not impact movement.7 They also indicate that sleep and anaesthesia greatly impair brain clearance rather than improve it.
With advancing age, several structural and functional changes compromise glymphatic efficiency. These include reduced aquaporin-4 (AQP4) polarization, diminished arterial pulsatility, and impaired cerebrospinal fluid and interstitial fluid exchange. Consequently, aged brains exhibit reduced clearance of Aβ, facilitating its aggregation into extracellular plaques, which is a defining feature of early AD pathology.8
Experimental evidence from aged rodent models and human neuroimaging studies has demonstrated a significant decline in glymphatic clearance, which correlates with an increased Aβ burden. Interventions that improve sleep architecture, enhance vascular function or stimulate AQP4 expression are being explored as therapeutic strategies to restore glymphatic function and slow AD progression.6
Microglial dysfunction and immune dysregulation in ageing
The brain’s macrophages, known as microglia, are essential for preserving the homeostasis of the central nervous system because they monitor the surroundings, control inflammatory reactions, remove debris, including Aβ, and promote synaptic plasticity.9
However, with ageing, microglia undergo functional decline, shifting from a neuroprotective to a pro-inflammatory state due to transcriptional changes that upregulate inflammatory genes while downregulating homeostatic genes.10 This dysfunction is exacerbated by reduced phagocytic efficiency, which impairs the clearance of Aβ and contributes to plaque accumulation, neurotoxicity and tau pathology.8
At the same time, pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, tumour necrosis factor-α and tumour necrosis factor-γ are elevated due to systemic immunological dysregulation in ageing, which impairs memory formation, synaptic integrity and neuronal function.11 Central to this process is the transcription factor nuclear factor kappa B (NF-κB), which is activated by oxidative stress, mitochondrial dysfunction and cellular senescence, which perpetuates inflammatory signalling. Together, microglial dysfunction and immune dysregulation create a vicious cycle of chronic inflammation and neuronal damage, accelerating age-related cognitive decline.12
Endoplasmic reticulum stress and apoptosis
The endoplasmic reticulum (ER) is crucial for protein folding, calcium homeostasis and lipid synthesis. Under normal conditions, the ER maintains proteostasis via a tightly regulated quality control system. With ageing, protein misfolding and metabolic stress increase, neurofibrillary tangles destabilize microtubules and p-tau is induced. Aβ is also produced through the overexpression of beta-site amyloid precursor protein-cleaving enzyme 1.13
A distinct initial imbalance or shock of aberrant proteins triggers apoptosis by activating caspases via both extrinsic and intrinsic (mitochondrial) pathways, which ultimately results in cell death.14 Through noradrenergic signalling, calcium dysregulation and oxidative stress, ageing makes neurons more susceptible to ER stress.15
Apolipoprotein E hypothesis
Apolipoprotein E (APOE) plays a role in the normal breakdown of lipoproteins and the transport of fats and fat-soluble substances to the lymphatic system. It is initially synthesized in the brain and liver. The three alleles of the APOE gene (ε2=8%, ε3=77% and ε4=15%) are found on chromosome 19. AD was also linked to APOE ε4.16 APOE ε4 disrupts lipid transport, impairs glymphatic clearance and promotes neuroinflammation, thereby accelerating Aβ/tau accumulation and synaptic dysfunction.16
Ageing alters the expression and function of APOE, leading to reduced lipid transport efficiency, impaired neuronal repair and increased vulnerability to neuroinflammation and Aβ accumulation, particularly in individuals carrying the APOE ε4 allele.17
Neurotrophic factor hypothesis
Brain-derived neurotrophic factor (BDNF) is essential for immune system lifespan, learning, memory and sleep.18 The action of BDNF on tropomyosin receptor kinase B sustains long-term potentiation. Neuronal degeneration and impaired memory formation can result from changes in tropomyosin receptor kinase B receptor levels or in BDNF expression.19 Ageing and AD reduce BDNF expression, weakening synaptic plasticity and memory-related neuronal survival pathways.18
Excitotoxic hypothesis
Glutamate, an excitatory neurotransmitter, is essential for memory and learning. It has both ionotropic (NMDA) and metabotropic receptors. AD is influenced by NMDA ionotropic glutamate receptors. During the resting membrane potential, magnesium ions block the voltage-dependent calcium channels of NMDA receptors. During depolarization, this barrier is removed, allowing calcium to enter the cell. The decrease in glutamate recycling in AD causes hyperexcitation of these receptors. These processes cause cell death and damage to neurons by increasing calcium influx.20
Age-related decline in glutamate recycling and mitochondrial buffering increases excitotoxic vulnerability, driving calcium-dependent neuronal death in AD.21
Therapeutic implications: Targeting ageing to treat Alzheimer’s disease
Traditional therapeutic approaches for AD have primarily focused on reducing the Aβ burden or preventing p-tau aggregation.Despite decades of effort, these strategies have yielded limited clinical success rates. In contrast, targeting upstream ageing mechanisms offers a new therapeutic paradigm, involving interventions that delay, prevent or even reverse AD-related processes. These interventions may include a variety of lifestyle adjustments and biological and medical therapies.
Lifestyle modifications
To maintain a normal weight and level of activity, lifestyle changes that incorporate physical activity, a balanced diet, restful sleep, stress management and mental activity are generally safe and have been demonstrated to have neuroprotective effects (Table 1).22–41
Table 1: Anti-ageing lifestyle interventions as therapeutic strategies for AD22–41
| Category | Mechanism | Effect on AD/ageing | Level of evidence | References |
| Physical exercise | Increases BDNF, IGF-1 and VEGF; enhances glymphatic clearance (AQP4-dependent); reduces inflammation (NF-κB inhibition) | Enhances neurogenesis, reduces Aβ/tau and improves cognition | Meta-analyses and human trials | 23–27 |
| Mediterranean/MIND diet | Rich in omega-3 fatty acids and polyphenols; reduces oxidative stress; modulates gut microbiota | Reduces Aβ aggregation, neuroinflammation and cognitive decline | Cohort studies and clinical trials | 28–30 |
| CR | Activates SIRT1 and AMPK; induces autophagy | Delays ageing, reduces Aβ plaques and improves mitochondrial function | Preclinical + early human studies | 31–34 |
| Sleep optimization | Supports glymphatic Aβ/tau clearance during slow-wave sleep | Prevents protein accumulation and reduces neuroinflammation | Observational and mechanistic studies | 35–37 |
| Stress management | Normalizes HPA axis; reduces cortisol; increases BDNF | Reduces hippocampal atrophy, Aβ production and neuroinflammation | Observational + small interventional trials | 38,39 |
| Cognitive engagement | Increases synaptic plasticity, hippocampal neurogenesis and BDNF | Strengthens cognitive reserve and slows Aβ/tau pathology | Epidemiological and longitudinal studies | 40,41 |
AD = Alzheimer’s disease; AMPK = adenosine monophosphate-activated protein kinase; AQP4 = aquaporin-4; Aβ = amyloid-beta; BDNF = brain-derived neurotrophic factor; CR = caloric restriction; HPA = hypothalamic–pituitary–adrenal; IGF-1 = insulin-like growth factor 1; MIND = Mediterranean–Dietary Approaches to Stop Hypertension Intervention for Neurodegenerative Delay; NF-κB = nuclear factor kappa B; VEGF = vascular endothelial growth factor.
Physical activity
The most efficient non-pharmacological strategy to affect brain ageing and AD risk is physical exercise, which includes morning gymnastics, walking, swimming, gardening, climbing stairs and housework. The WHO recommends 150 min/week of moderate activity. Through the following processes, combined exercise training – including aerobic, strength, balance and coordination training — as well as cognitive and social activities, appears to offer significant benefits for those with AD.22
First, by upregulating BDNF, insulin-like growth factor 1 (IGF-1) and vascular endothelial growth factor (VEGF), exercise antagonists alter AD by improving neuroplasticity and expanding hippocampus volume.23 These elements play a role in normal hippocampal regeneration and angiogenesis (regulated by IGF-1 and VEGF), as well as learning (regulated by IGF-1 and BDNF).22 It also enhances glymphatic clearance, promoting Aβ removal via AQP4-dependent mechanisms, which is particularly critical in APOE ε4 carriers.24 BDNF depletion reduces TrkB-mediated synaptic resilience and exacerbates glutamate excitotoxicity. Exercise-induced BDNF upregulation may counteract this by enhancing calcium buffering.22
Second, it improves mitochondrial biogenesis and glucose metabolism, which counteracts cerebral hypometabolism.25 Finally, it exerts anti-inflammatory, anti-immunity, antioxidant, hippocampal insulin signalling, autophagy and gut microbial ecosystem.26 Gamma-aminobutyric acid, irisin, dopamine and other chemicals are involved in these mechanisms.27
Exercise mimetics aim to replicate the cellular benefits of physical activity, such as mitochondrial biogenesis and insulin sensitivity, without physical exertion. However, they lack the full systemic effects of real exercise and may raise safety concerns with chronic use.
By promoting mitochondrial biogenesis and improving glucose metabolism, 5′ adenosine monophosphate-activated protein kinase (AMPK) activator lowers Aβ accumulation and enhances brain energy balance.42 Therapeutic strategies for APOE ε4 carriers focus on lipid-targeted approaches such as peroxisome proliferator-activated receptor delta agonists (e.g. GW501516), which may mitigate impaired transport and reduce neuroinflammation.43
However, despite the numerous mechanisms, further research is necessary to elucidate the exercise variables (including kind, volume, duration and intensity) that influence AD.
Nutrition
Eating a well-balanced diet rich in fruits, vegetables, whole grains, lean meats and healthy fats might help prevent insulin resistance, gut–brain axis dysfunction and oxidative stress neuroinflammation, in addition to offering essential nutrients for neuroprotection.5
Delays in cognitive decline and a lower risk of AD are linked to nutritional strategies such as the Mediterranean diet, the Dietary Approaches to Stop Hypertension (DASH) diet, the Mediterranean–DASH Intervention for Neurodegenerative Delay and the Stop Hypertension (GUSTO) diet.26,29 These diets are high in fibre, plant-based polyphenols, omega-3 fatty acids and vitamins E and B12, and low in saturated fats and refined sugars, all of which modulate the hallmarks of ageing and neurodegeneration.28,30 Omega-3 (polyunsaturated fatty acid) is integral to neuronal membrane integrity and has been shown to reduce Aβ aggregation, suppress pro-inflammatory cytokine production, promote synaptic plasticity and reduce the risk of cognitive decline.28 Royal jelly provides neuroprotection against tau and Aβ aggregation, especially with ageing through synaptic signal transduction, antioxidant system enhancement, inflammation suppression and increased neurotrophin production – all of which support normal neuronal structure and function.31
Dysbiosis with ageing is linked to increased permeability of the gut and BBB, promoting systemic inflammation and AD pathology – a process that can be attenuated by probiotic, prebiotic or faecal microbiota transplantation.30
Reducing caloric intake by 20–60% without causing starvation is known as calorie restriction (CR). Mice’s lifespan can be increased by up to 40% or more by delaying age-related diseases.31 The degree of CR, the length of the diet, the age, sex and general health status of the individual all affect how CR works.32
Mechanisms that directly prevent ageing-related AD pathogenesis include SIRT1 gene activation, enhanced insulin sensitivity and autophagy induction.33 CR combined with intermittent fasting is more effective in improving cognitive function and reducing Aβ accumulation.34
CR mimetics are substances that, without actually lowering caloric intake, attempt to replicate the physiological effects of CR. Natural substances, including resveratrol, curcumin and quercetin, as well as pharmaceutical medications such as metformin, glucagon-like peptide-1 receptor agonists, rapamycin and spermidine, are examples of CR mimetics.44
Resveratrol, a natural polyphenolic chemical present in vegetables and fruits, has been shown to combat inflammation, oxidative stress, apoptosis, synaptic dysfunction, mitochondrial dysfunction and angiogenesis.42,44 It also reduces Aβ aggregation, inhibits p-tau levels and enhances mitochondrial biogenesis. Clinical evidence for resveratrol in patients with AD is mixed; while it has a high safety profile, its efficacy is considered partial or trending. Further data are needed to validate its treatment potential for AD, suggesting a need for tempered enthusiasm.42 It has both physical activity and CR mimetics.
Metformin, a widely used AMPK activator for type 2 diabetes, promotes autophagy, reduces oxidative stress and improves insulin sensitivity in the brain, counteracting insulin resistance often observed in early AD.45 Some studies suggest that metformin reduces p-tau and neuroinflammation in animal models of AD.32
Rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, delays brain ageing by enhancing autophagy and lysosomal clearance of damaged proteins, including Aβ and p-tau. It also reduces microglial activation and inflammaging, which are exacerbated by age and contribute to cognitive decline.44
Spermidine, a polyamine with CR-mimetic properties, increases autophagy and exerts neuroprotective effects in AD models by decreasing tau fibrillation and Aβ burden, boosting mitochondrial function and improving memory.46
Curcumin, the principal polyphenol in turmeric, exhibits anti-Aβ activity, antioxidant properties, anti-inflammatory effects, antiapoptotic functions and modulation of cellular pathways through epigenetic processes. It binds to Aβ plaques, inhibits aggregation, promotes clearance and modulates microglial activity.47
Combinatorial methods can be used to increase curcumin’s therapeutic effectiveness. For example, curcumin and ascorbic acid together increase the anti-inflammatory response.48
Quercetin, a natural flavonoid found in fruits and vegetables, has antioxidant and anti-inflammatory effects.49 It produces its anti-inflammatory effects via activation of AMPK, which inhibits the activation of pro-inflammatory pathways. It also limits the damaging effects of inflammation on neuronal cells and improves mitochondrial function in mice with AD.50 Quercetin also lowers Aβ aggregation, p-tau phosphorylation and microglial activation. It restores acetylcholine levels through the inhibition of the hydrolysis of acetylcholine by the acetylcholinesterase enzyme.51
Genistein, a soy-derived isoflavone, mimics the oestrogenic effects of oestrogen, providing neuroprotection in postmenopausal women. It has multimodal properties, including antioxidant, anti-inflammatory, anti-amyloidogenic, anti-gut dysbiosis, pro-autophagy actions and augmentation of neural plasticity.52
Blueberries, blackberries and purple corn contain anthocyanins that have anti-inflammatory, anti-apoptotic, antioxidant, neuroprotective and cholinergic neurotransmission properties. They also reduce oxidative stress and decrease Aβ aggregation. They enhance synaptic signalling, improve spatial working memory and reduce tau pathology.53 The metabolic rate of anthocyanins varies among individuals, potentially affecting their overall effectiveness.54
Sleep
Sleep disruption affects the glymphatic system during slow-wave sleep, reducing the clearance of Aβ and p-tau.35 It also increases pro-inflammatory cytokine levels, triggers ER stress and disrupts synaptic homeostasis by reducing BDNF expression, leading to neurodegeneration and pathological progression.36 Thus, glymphatic mechanisms remain contested. Ageing and AD disproportionately reduce slow-wave sleep and create a vicious cycle of neurodegeneration.37
Stress management
Chronic psychological stress contributes to AD pathogenesis via neuroendocrine, inflammatory and structural mechanisms. By disrupting the hypothalamic–pituitary–adrenal (HPA) axis, ageing intensifies these effects by raising cortisol levels, which impair long-term potentiation, diminish neurogenesis and cause hippocampal atrophy – all of which are critical for memory.38 Additionally, glucocorticoids stimulate Aβ production, enhance p-tau and impair Aβ clearance while activating NF-κB-mediated neuroinflammation and microglial dysfunction, thereby contributing to inflammaging and neuronal death.39
It has been demonstrated that stress management techniques, including yoga, mindfulness and cognitive behavioural therapy, raise BDNF expression, increase hippocampus volume and return cortisol levels to normal.38
Mental activity
Maintaining intellectual and mental activities can be achieved by regularly participating in activities, such as crossword puzzles, education, reading books and magazines, puzzle organization, playing games like chess, board games, checkers and cards and by taking music courses. These activities aid in enhancing visual memory, learning capacity, logical reasoning, attention, focus and perceptiveness.27
Mechanistically, cognitive engagement enhances synaptic density, promotes dendritic complexity, upregulates BDNF and supports hippocampal neurogenesis, creating structural resilience against neurodegeneration.40 Cognitive enrichment has been demonstrated to lower Aβ burden and decrease functional decline in both human and animal models, including APOE ε4 carriers.26
Therefore, the interventions such as exercise, cognitive stimulation and dietary polyphenols aim to restore BDNF activity and improve synaptic resilience.
Sociostation
Ageing often leads to social isolation, comparatively more isolation and loneliness. These psychosocial deficits exacerbate HPA axis dysregulation, elevate cortisol levels, induce hippocampal atrophy and intensify neuroinflammation, all of which contribute to accelerated neurodegeneration and Aβ/p-tau pathology.41
Conversely, structured social programmes – such as group therapy and intergenerational initiatives – improve mood and memory, reduce Aβ deposition, elevate BDNF expression, preserve long-term potentiation and delay functional decline in patients with AD.55
Creativity
Mechanistically, creative interventions such as music, art and dance enhance emotional regulation, support cognitive performance and boost levels of BDNF and nerve growth factor in patients with AD. These effects are mediated by reduced neuroinflammation and stress pathways.56
Medical therapies
Acupuncture
As acupuncture improves neuroinflammation, synaptic plasticity, nerve cell death and the brain’s synthesis and aggregation of Aβ, it can help with memory and cognitive impairment in patients with AD (Table 2). It is considered non-invasive, generally safe and associated with improved cognitive markers in small-scale trials.57
Cellular senescence
It is defined as the irreversible growth arrest of damaged or stressed cells, which contributes significantly to age-related neurodegeneration through the release of the senescence-associated secretory phenotype (SASP) – a mixture of chemokines, proteolytic enzymes and cytokines.58 While senolytics reduce pathology in mice, translation is limited. The SToMP-AD pilot (dasatinib + quercetin) showed safety but no cognitive benefit, highlighting quercetin’s poor CNS penetration.59
On the other hand, senomorphics such as rapamycin and metformin suppress the harmful SASP without inducing cell death and have shown efficacy in reducing tau pathology and modulating neuroinflammation in preclinical settings.44 There is some debate on the usefulness of senescence, despite its potential for treatment. Their inability to specifically eradicate senescent cells is the first drawback. Second, while senolytics may be less effective if used later, administering them too soon causes stem cell depletion, which speeds up the ageing process and causes thrombocytopenia in the elderly. The type and quantity of senescent cells that should be eliminated for the best results are also controversial.59 These approaches remain experimental with limited human data.
Epigenetic ageing and reprogramming
Age-related epigenetic changes, such as DNA methylation drift and histone modification alterations, can silence key neuroplasticity-related genes and are predictive of AD risk. Epigenetic clocks, especially the Horvath clock, serve as biomarkers of biological ageing and correlate more closely with AD pathology than with chronological age.60
Histone deacetylase inhibitors, such as sodium butyrate, have been shown to restore histone acetylation, increase BDNF expression and improve memory performance in AD models.61 More recently, partial epigenetic reprogramming using Yamanaka factors has demonstrated reversal of age-associated DNA methylation patterns, reduced Aβ pathology and enhanced cognitive function in murine models without inducing tumourigenesis.62
Enhancing autophagy and proteostasis
Autophagy declines with age, leading to toxic protein accumulation. mTOR inhibition by rapamycin restores autophagy and reduces Aβ/p-tau pathology, thereby improving cognition.52 Natural compounds such as spermidine, curcumin and resveratrol also induce autophagy and show cognitive benefits.42 Without adversely affecting other proteins such as the Notch receptor or amyloid precursor-like protein 1, the autophagy-activator small-molecule enhancer of rapamycin-28 promotes the degradation of Aβ and the C-terminal segment of the amyloid precursor protein.52
Hormonal therapy
Neuroprotective substances include IGF-2, growth hormone-releasing hormone, gonadotropin-releasing hormone and oestrogen. Gonadotropin-releasing hormone slows down ageing and encourages neurogenesis in mice.3 In AD models, IGF-2 improves cognition by promoting neurogenesis and synaptogenesis.63 Additionally, it regulates transcription factors, including nuclear factor erythroid 2-related factor 2 and NF-κB, that are connected to inflammation and oxidative stress.64
Biological therapies
In addition to repairing nerve damage in AD, neural stem cells can also slow down ageing and neurodegenerative disorders because of their capacity to develop into neurons, astrocytes and oligodendrocytes. They have been demonstrated to improve pathogenic events and behaviours in AD mice and to reduce neuroinflammation, synaptic dysfunction and metabolic dysfunction.65 Furthermore, enkephalinase (neprilysin), which is secreted by adipose-derived stem cells, can directly break down Aβ plaques (see Table 3).30,42,59,61,66–73
Table 3: Successes versus failures in geroscience-based AD trials30,42,59,61,66–72
| Intervention | Mechanism | Clinical outcome | Notes/references |
| Aducanumab | Monoclonal antibody targeting amyloid-β plaques | FDA-approved (2021); limited clinical benefit observed; approval process controversial | Despite amyloid clearance, efficacy in slowing cognitive decline was minimal; approval was contentious due to questionable clinical significance68 |
| Donanemab | Monoclonal antibody targeting modified amyloid-β plaques | Positive results in early Alzheimer’s stages; reduced amyloid burden and slowed cognitive decline | Demonstrated efficacy in early-stage AD; concerns remain regarding side effects and long-term benefits69 |
| Crenezumab | Monoclonal antibody targeting amyloid-β plaques | Mixed results; no significant cognitive improvement in moderate AD; some benefit in mild cases | Showed potential in early-stage AD but failed to meet primary endpoints in moderate AD70 |
| Solanezumab | Monoclonal antibody targeting soluble amyloid-β | Failed to slow cognitive decline in mild and preclinical AD stages | Despite initial optimism, did not demonstrate efficacy in large-scale trials71 |
| Senolytics (dasatinib + quercetin) | Clears senescent cells | Preclinical success; on-going phase II trials in AD | Early studies in mice showed reduction of neuroinflammation and improved cognition59 |
| CR mimetics (resveratrol) | Activates SIRT1, mimics caloric restriction | Phase III trials on-going; mixed results in earlier stages | Evidence of cognitive benefits exists, but larger trials are needed42 |
| Epigenetic modulators (sodium butyrate) | Increases histone acetylation, BDNF levels | Preclinical success; early-phase human trials under way | Demonstrated memory improvement in animal models; human data are limited61 |
| Epigenetic reprogramming (Yamanaka factors) | Resets DNA methylation patterns | Preclinical success in mice; not yet tested in humans | Reversed age-related cognitive decline in animal models; human applicability remains untested61 |
| Stem cell therapy (neural stem cells) | Secretes neurotrophic factors | Preclinical success; animal studies indicate potential amyloid reduction | Early studies suggest neuroprotection and cognitive enhancement; human trials are in early stages72 |
| Young plasma exchange | Transfers rejuvenating factors from young plasma | Pilot trials show safety; potential efficacy in reversing age-related cognitive decline | Early-phase trials indicate safety; efficacy data remain preliminary66 |
| Faecal microbiota transplantation | Modulates gut microbiota | Reversed ageing-related cognitive decline in mice; limited human trials | Animal studies show promise in reversing cognitive decline; human data are scarce30 |
AD = Alzheimer’s disease; BDNF = brain-derived neurotrophic factor; FDA = Food and Drug Administration.
Young bone marrow transplantation helps older mice maintain synaptic connections, cytokine levels and cognitive symptoms, extending their maximum lifespan by 30%.74 However, the practical use of bone marrow transplantation is restricted by transplant rejection and the scarcity of young bone marrow donors.73
In addition to reducing soluble Aβ levels in the blood and Aβ plaques in the brains of aged rats, whole-blood replacement dramatically improves spatial memory.75 Delivering a wider variety of anti-ageing agents that target several ageing markers simultaneously is made possible by blood rejuvenation. Although it necessitates an adequate blood supply and may result in negative reactions, this strategy might be more effective.4 Further study into long-term plasma therapy is necessary, as evidenced by encouraging outcomes in patients with AD who received 4 weekly infusions of young fresh-frozen plasma.66
When the gut microbiota of young mice is transplanted into older animals, immunosenescence and neuroinflammation are reversed, and hippocampal neurogenesis, behaviour and cognition are all improved (Table 2).42,59,61,62,65–67
Table 2: Anti-ageing pharmacological and biological therapies as the therapeutic strategies for AD42,59,61,62,65–67
| Category | Intervention | Mechanism | Stage of evidence | Clinical trial status |
| Senolytics | Dasatinib + quercetin–fisetin | Clears senescent microglia | Dasatinib + quercetin (phase II)–fisetin (phase I)59 | On-going/early-phase trials |
| CR mimetics | Resveratrol | Activates SIRT1, autophagy | Phase III human trials42 | Completed phase III trials |
| Epigenetic modulators | Sodium butyrate | ↑ Histone acetylation, BDNF | Preclinical, improves memory61 | Not yet in clinical trials |
| Epigenetic reprogramming | Yamanaka factors | Resets DNA methylation | Preclinical (mice)62 | Not yet in clinical trials |
| Stem cells | Neural stem cells | Secretes neurotrophic factors | Mouse models show Aβ reduction65 | Preclinical/animal studies |
| Young plasma | Plasma exchange | Rejuvenates systemic factors | Pilot trials show safety66 | Early-phase clinical trials |
| Gut microbiota | Faecal transplant | Reduces neuroinflammation | Reversed ageing in mice67 | Preclinical/animal studies |
AD = Alzheimer’s disease; Aβ = amyloid-beta; BDNF = brain-derived neurotrophic factor; CR = caloric restriction; SIRT1 = sirtuin 1.
Despite encouraging preclinical data, challenges remain. Ethical concerns, risk of tumourigenesis, immune rejection and low survival rates post-transplantation must be addressed before clinical translation.
Other possible therapies
Nonsteroidal anti-inflammatory drugs such as ibuprofen; antiviral medications such as valacyclovir; some antibiotics; mitochondrial function regulators such as nilotinib; metabolic activators such as l-serine, N-acetyl cysteine, nicotinamide riboside and l-carnitine tartrate; neural repair medications such as CT1812 and simufilam; and antioxidants have been shown to have neuroprotective effects and are being investigated for the treatment of AD.4 Management of comorbidities – particularly vascular risk factors (hypertension, diabetes, atrial fibrillation) and sensory deficits such as hearing loss – is crucial to reduce additive contributions to AD pathology. These represent modifiable accelerators of ageing-related neurodegeneration.
Translational challenges
Although many geroscience-based strategies show promise in animal models, translation to humans remains limited. For example, senolytic combinations such as dasatinib + quercetin reduced Aβ and tau pathology in mice but showed only safety, without cognitive benefits, in the phase II SToMP-AD (Phase II Clinical Trial to Evaluate the Safety and Feasibility of Senolytic Therapy in Alzheimer’s Disease; ClinicalTrials.gov identifier: NCT04685590) pilot.4 Similarly, the pioglitazone TOMMORROW trial (A Double Blind, Randomized, Placebo Controlled, Parallel Group Study to Simultaneously Qualify a Biomarker Algorithm for Prognosis of Risk of Developing Mild Cognitive Impairment Due to Alzheimer’s Disease [MCI Due to AD] and to Test the Safety and Efficacy of Pioglitazone [AD-4833 SR 0.8 mg QD] to Delay the Onset of MCI Due to AD in Cognitively Normal Subjects; ClinicalTrials.gov identifier: NCT01931566) was halted after failing to meet efficacy endpoints, despite a strong preclinical rationale.76 Such outcomes underscore the disconnect between preclinical success and human disease, driven by species differences, trial design limitations and the heterogeneity of late-onset AD.77 Acknowledging these translational barriers is critical for developing predictive models and biomarker-guided trials that can better capture the complexity of AD.
Ethical considerations
The development of ageing-targeted therapies for AD raises important ethical challenges that must be addressed alongside scientific progress. One central issue is the difficulty of obtaining truly informed consent from patients with cognitive impairment, who may have limited decision-making capacity and require careful involvement of caregivers or legal representatives.78 Equity concerns also arise, as access to costly and experimental biologics, such as stem cell therapies or plasma exchange, may be restricted to privileged populations, potentially widening existing health disparities.72 Finally, researchers and clinicians have a responsibility to communicate cautiously about unproven interventions to avoid fostering unrealistic expectations or offering false hope to vulnerable patients and families.79 A balanced and transparent ethical framework is essential to guide the responsible translation of geroscience-based therapies into clinical practice.
Limitations
This article proposes that treating AD by targeting the biological mechanisms of ageing itself (a geroscience approach) is a promising new strategy. Interventions such as senolytics, CR mimetics and lifestyle changes show potential by addressing the root causes of AD. However, this approach faces significant limitations. First, there is a major gap between successful animal studies and proven human treatments, with many therapies still in early trials whose long-term safety remains unknown. Second, as a narrative review, this analysis may be subject to author bias and does not systematically evaluate all evidence. Furthermore, ageing is highly individual, meaning personalized approaches based on genetics and biomarkers will be crucial, as a single universal treatment is unlikely to work for everyone. Finally, while safe, the effectiveness of lifestyle changes can be difficult to measure due to challenges with patient adherence and defining optimal implementation.
Future directions and conclusion
AD can be viewed not just as a neurodegenerative condition but as a manifestation of accelerated brain ageing. By targeting the root causes of ageing through a geroscience framework, we have the opportunity to delay or even prevent AD onset. Rather than targeting downstream symptoms alone, emerging strategies seek to intervene earlier in the disease course by addressing the biological ageing mechanisms that drive AD pathogenesis. Future research should focus on integrated, multimodal interventions that combine lifestyle modification with pharmacological and biological therapies. Tailored approaches – based on genetic risk profiles (e.g. APOE status), comorbidities and individual ageing trajectories – may optimize clinical outcomes. To evaluate the long-term safety and effectiveness of innovative treatments such as senolytics, epigenetic modulators and stem cell-based therapies in older populations, extensive, longitudinal clinical trials are also required. Developments in biological age biomarkers, machine learning and systems biology have the potential to improve risk assessment and therapy customization.











