
Parkinson’s disease (PD) is a neurological disorder characterized by a progressive degeneration of dopaminergic neurons located in the substantia nigra pars compacta (SNc).1 Dopaminergic neurons of the SNc send their axons along the nigrostriatal pathway to the striatum, where dopamine (DA) acts to modulate post-synaptic signaling. The striatum comprises a heterogeneous population of neurons that receives input from a number of regions, including motor and sensory cortices, and under the influence of DA utilizes this information to guide motor behavior. It is a substantial loss (>80%) of this DA innervation that is responsible for the classic triad of PD symptoms, i.e. resting tremor, muscular rigidity, and bradykinesia.
Since the primary pathological consequences of PD are due to the degeneration of a specific set of neurons that specifically release the neurotransmitter DA, it was suggested that DA-replacement therapy may be effective in treating the disease.2 DA itself is not a suitable candidate since it does not cross the blood–brain barrier. For this reason, the pharmacological treatment of PD is largely based on administration of the precursor of DA, 3,4-dihydroxy-L-phenylalanine (L-DOPA). This treatment remains the most effective way to reduce the major symptoms of the disease; however, with prolonged administration the development of debilitating involuntary movements, known as L-DOPA-induced dyskinesias (LIDs), occurs.
In this article, we will present some background information about the regulation of striatal neuron activity and how this is altered not only in the DA-depleted state, but also following chronic L-DOPA administration. Such information is important in guiding more effective treatment strategies for PD.
Dopamine Regulation of Striatal Output
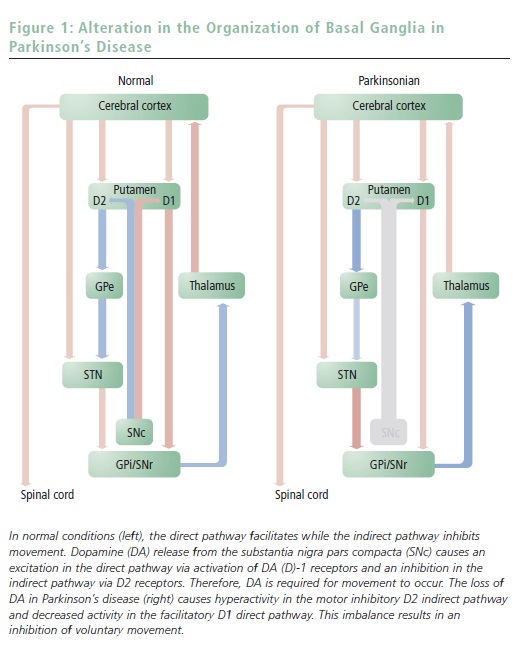
Two distinct pathways connect the striatum to the basal ganglia ‘output’ structures (see Figure 1). The so-called type I medium-spiny neurons (MSNs) directly inhibit the internal segment of the globus pallidus (GPi) and the substantia nigra pars reticulata (SNr). Thus, in this ‘direct pathway’ striatal neurons disinhibit the thalamic neurons responsible for excitation of the pre-motor cortex.3 As such, activation of the direct pathway facilitates voluntary movements.
The other group of MSNs, known as type II neurons, inhibits the external segment of the globus pallidus (GPe), which suppresses the excitatory cells of the subthalamic nucleus (STN). STN neurons are glutamatergic and excitatory and project to the GPi/SNr. This circuitous route to the thalamus is known as the ‘indirect pathway’ and provides a mechanism to modulate the disinhibitory actions of the direct pathway on the thalamus. Thus, the direct and indirect pathways act in concert to regulate motor control handled by the basal ganglia. The direct pathway facilitates movement by decreasing the tonic inhibition of basal ganglia ‘outputs’ to the thalamus, while the indirect pathway suppresses movements by increasing the inhibitory pathway.
In addition to being anatomically divergent, the direct and indirect pathways are also neurochemically distinct. Specifically, MSNs of the direct pathway express high levels of DA (D)-1 receptors (type I neurons), whereas neurons that form the indirect pathway preferentially express the D2 receptors (type II neurons).4,5 Thus, DA release in the striatum differentially regulates the output of the direct and indirect pathways. D2-receptor stimulation inhibits type II striatal neurons that comprise the indirect output pathway, whereas D1-receptor stimulation facilitates activity in type I (direct pathway) striatal neurons. As such, the actions of DA on the direct and indirect pathways are antagonistic, and given that the direct and indirect pathways excite and attenuate movement, respectively, the net effect of DA is a facilitation of motor output (see Figure 1).
Dopamine Regulation of Striatal Output in
Parkinson’s Disease
As mentioned earlier, PD symptoms are associated with DA neuron degradation; however, this system is relatively resilient to decreases in SNc neuron number. Indeed, during the early stages of the disease, compensatory changes such as a decrease in DA inactivation due to a loss of the DA transporter, an increase in D2 receptors, and an increase in DA synthesis in the remaining DA terminals can adequately compensate for the decreased SNc neuron number. However, once the progressive degeneration of DA neurons results in >80% neuronal loss, these mechanisms are no longer adequate to compensate for the diminished DA release in the striatum. Given the opposing role for D1 and D2 receptors in the striatal outputs, this decrease in DA levels leads to hyperactivity within the type II (D2-containing) MSNs and a hypoactivity of type I (D1-containing) MSNs, resulting in an imbalance between the direct and indirect output pathways. As a consequence, an abnormal hyperactivity of STN and GPi emerges; this aberrant regulation results in a high inhibition of thalamic activity and the subsequent expression of parkinsonian motor impairment (see Figure 1).6
Dopamine Regulation of Striatal Output Following L-DOPA
At present, replacement therapy with a precursor of DA, L-DOPA, is the most effective therapeutic approach in treating the symptoms of PD.7 This treatment effectively restores terminal DA levels in the striatum and re-establishes the balance between activation of the direct pathway (increased by activation of D1 receptors) and the indirect pathway (attenuated by activation of D2 receptors). Thus, L-DOPA pharmacotherapy is highly effective in reducing symptoms of PD on a short-term basis. Nonetheless, after three to five years of treatment, motor fluctuations, loss of efficacy, and development of involuntary movements known as LIDs occur, significantly limiting the usefulness of L-DOPA as a therapeutic tool. Despite the clinical importance of this side effect, relatively little is known about the mechanisms underlying the generation of LIDs.8

One avenue of research has proposed that LIDs are due to an aberrant form of cortico-striatal synaptic plasticity.9–11 Thus, in addition to the impairment of dopaminergic modulation of striatal output, the loss of DA neurons in PD is proposed to attenuate striatal cellular learning known as long-term potentiation (LTP) and long-term depression (LTD).12,13 Under ‘normal’ conditions, transient high-frequency stimulation (HFS) of motor cortical afferents to the striatum significantly attenuates subsequent responses to motor-cortex activation in both direct and indirect output neurons (see Figure 2). However, in an animal model of PD (6-OHDA lesion), HFS still evoked LTD in direct (D1-containing) neurons while having no observable effect on indirect (D2-containing) neurons. This decreased adaptive response to motor-cortical activation is consistent with the reported dependence of LTD on D2-receptors stimulation.14
Thus, a decrease in DA levels within the striatum would result in the loss of a component essential for the development of LTD in type II neurons and a loss of control over motor inhibition. Following chronic L-DOPA
administration, the ability of type I output neurons to demonstrate LTD in response to motor cortical HFS was unchanged. However, type II neurons demonstrated the opposite plastic response, i.e. a potentiation of synaptic transmission (see Figure 2). Such a dramatic alteration in information processing within the striatum will have drastic effects on the ability of this region to regulate motor behavior. Furthermore, although acute L-DOPA administration should restore the balance between the direct and indirect pathways following chronic administration, an augmented indirect pathway response to sustained motor cortical activation would result in an imbalance between the two output pathways in response to acute administration of L-DOPA. Therefore, with repeated L-DOPA administration, the consequential alteration in synaptic plasticity in the direct versus indirect output pathways of the dorsal striatum may underlie the loss of inhibitory motor control and the resultant dyskinesias induced by L-DOPA (see Figure 3).
Summary and Hypothesis
The cortico-striatal system is regulated in a complex manner by multiple feedback and feed-forward systems that have a high capacity to compensate for a large degree of pathological insult. However, it may be these same compensatory processes that are responsible for the aberrant involuntary movements observed following chronic L-DOPA treatment. Thus, although L-DOPA pharmacotherapy is highly effective in reducing the classic triad of the symptoms of PD on a short-term basis, chronic L-DOPA administration results in altered cortico-striatal plasticity, significantly altering the balance between direct and indirect output pathways. We suggest that these plastic changes underlie the development of L-DOPAinduced dyskinesias. Given that continuous DA-receptor stimulation is known to circumvent DA-induced plasticity,15,16 it is proposed that a similar type of continuous DA stimulation could treat Parkinson’s pathology without induction of dyskinesias.17 Such information is important in guiding future pharmacotherapy or treatment regimens to counteract this debilitating side effect in the treatment of PD.











