Multiple sclerosis (MS) is an inflammatory disease of the central nervous system affecting almost 3 million individuals worldwide.1–3 The precise pathogenesis of the disease is unknown, though migration of peripheral T and B lymphocytes into the central nervous system is considered to be one of the major pathophysiologic mechanisms.4–6 Sphingosine-1-phosphate (S1P) modulators, through induction of S1P receptor (S1PR) down-regulation, prevent lymphocyte egress from lymphoid tissue.7,8 The US Food and Drug Administration’s (FDA) approval of the first S1PR modulator for use in MS, fingolimod, in 2010, was followed in 2018 by approval in patients with MS aged 10–18 years following a randomised controlled trial showing efficacy of this agent over interferon (IFN) beta-1a.9
Despite studies showing efficacy of fingolimod in MS, phase II and III, and long-term follow-up studies revealed significant adverse events, of which cardiac events were of particular concern. Reasons for this are related to its non-selective binding to several S1PRs: S1PR1 and S1PR3–5, which include receptors found on cardiac tissue.5,10–12 Noted side effects include significant
first-dose bradycardia, second- and third-degree atrioventricular block, QT interval prolongation, hypertension, macular oedema and pulmonary toxicity.9,10,13 Multiple agents with selective S1P inhibition have therefore been developed to address these safety concerns.14,15 In this article we will review the biological basis for, and pivotal clinical trials of, one such agent, ozanimod.
Mechanism of action – sphingosine-1-phosphate receptors
S1P is a phospholipid-soluble signalling molecule that interacts with G protein-coupled receptors.7,16,17 In the decade between 1990 and 2000, five subtypes of S1PR were identified by scientists. Further studies demonstrated that S1PR1 took part in lymphocyte migration and was also found in cardiovascular structures, while S1PR2 modulated cardiovascular function, bone structure and fertility, together with S1PR3.18–21 Overall, S1PR1–3 have been postulated to be associated with cancer and metastases as they have a role in cell migration; and S1PR4 and S1PR5 were found to modulate migration of haematopoietic cells and lymphocytes, respectively.22 Antagonism of the S1PR1 was shown to decrease the number of circulating lymphocytes, leaving them sequestered in the lymph nodes.11,17
To benefit from this effect in the treatment of immune-mediated disorders such as MS and ulcerative colitis, pharmacological agents with targeted S1PR1 modulatory effects have been developed, including ozanimod, siponimod and ponesimod. These agents are at various stages of development. Siponimod, a selective S1PR1 and S1PR5 modulator, recently received FDA approval for use in clinically isolated syndromes, relapsing remitting MS (RRMS), and secondary progressive MS with active disease following demonstration of a 21% risk reduction in time to 3-month confirmed disability progression compared with placebo in the double-blind, phase III EXPAND trial.23 Ponesimod, a selective S1PR1 modulator, has been shown to decrease the number of contrast enhancing lesions on magnetic resonance imaging (MRI) in RRMS, and is currently in a phase III trial.24 Ozanimod, or RPC1063 – the subject of this review – is a small-molecule S1PR modulator with greater selectivity for the S1PR1 and S1PR5 subtypes.17,25
Overview of pharmacokinetics and pharmacodynamics
Animal studies evaluating the pharmacokinetics of ozanimod – including mouse and rat experiments – have established the presence of a high volume of distribution with an effective brain:blood ratio of 10:1 in mice and 16:1 in rats, and a half-life of 4.7 and 5.1 hours in mice and rats, respectively.26 In rodents, ozanimod decreased circulating T and B220+ lymphocytes by about 80% at 6 hours after dosing C57BL/6 mice (n=3, maximal blood concentration 159 nM) with 1.0 mg/kg and Sprague Dawley rats (n=6, maximal blood concentration 45 nM) with 0.5 mg/kg of RPC1063. Flow cytometry analysis of lymphocyte subsets showed that the reduction was selective to CD4+ CCR7+ and CD8+ CCR7+ T cells and significantly less on CCR7– lymphocytes. In vitro analysis showed a greater selectivity for the S1PR1 than S1PR2–4, and a comparable potency on S1PR1 relative to fingolimod (FTY720) and siponimod (BAF312).26
Ozanimod has one major (CC112273) and two minor (RP101988, RP101075) active metabolites.25 Limited literature available on these metabolites suggests that ozanimod may have similar pharmacokinetic properties to these metabolites. In 2018, Tran et al. reported that with
1 mg dosing, Cmax was 205 pg/mL for ozanimod, 354 pg/mL for RP101988 and 63.7 pg/mL for RP101075, with a stable Tmax of 6 hours with all metabolites. The elimination half-life was comparable between ozanimod and its metabolites, ranging between 19 and 22 hours.25 However, this study failed to identify the major metabolite of ozanimod: CC112273. No published reports of CC112273 are available, but in an abstract at the 2018 American Academy of Neurology (AAN) Meeting, CC112273 was reported to possess receptor potency and selectivity properties similar to those of ozanimod.27 Its elimination half-life has been reported to be 10–13 days, which is longer compared with fingolimod’s (8 days) and siponimod’s (56 hours) elimination half-lives.28,29
In a single-centre, randomised, double-blind, placebo-controlled phase I trial of 88 healthy volunteers, single oral ascending (administered at 0.3 mg, then 1.0 mg, 2.0 mg, 3.0 mg daily for 7 days) and multiple oral ascending (administered at 0.3 mg, 1.0 mg, 1.5 mg or 2.0 mg daily for 7 or 28 days) doses of ozanimod were investigated.17 Subjects reached plasma steady state concentration by day 7 with 0.3 and 1.0 mg dosing. The elimination half-life of ozanimod was 17.7 hours (standard deviation 3.14) with 1.0 mg dosing and ranged between 15 and 21 hours with the different dosing schemes tested in the study. This study confirmed the high volume of distribution seen in animal studies:26 volume of distribution ranged between 73 and 102 L/kg among study groups of 0.3–3.0 mg/day ozanimod dosing over 7–28 days.17
In terms of pharmacodynamics, Tran et al. showed that the absolute lymphocyte count (ALC) decreased by up to 68% in a dose dependent manner with administration of ozanimod 0.3, 1.0 and 2.0 mg for 7 consecutive days.17 Only after day 40, did the mean change in ALC from baseline decrease to <40%. Of note, in the dose-escalation cohort, ALC reached a nadir at 24 hours after reaching the goal dose, with a decrease of 67.4%, and did not return to baseline levels. The ALC appeared to plateau with a decrease of approximately 40% at day 7 post-dosing. Reduction in ALC was specific to the CCR7+ lymphocytes, with variable effect on CCR7– lymphocytes (no reduction in CCR7– 45RA+ CD4+, CCR7– 45RA+ CD8+ or natural killer cells).17
Clinical efficacy
Phase II
One randomised, double-blind, placebo-controlled, phase II trial of ozanimod was performed – RADIANCE (ClinicalTrials.gov identifier: NCT01628393). This study enrolled 258 subjects and was halted early at 6 months to initiate a phase III study upon achievement of the primary study endpoint (greater reduction in the total number of gadolinium-enhancing [GdE] lesions on MRI from week 12–24 with ozanimod 0.5 mg/day and 1.0 mg/day dosages compared with placebo).30
Participants in the RADIANCE study were randomised to three groups (placebo, ozanimod 0.5 mg and ozanimod 1.0 mg) in a 1:1:1 distribution. Although the groups were well-balanced with regards to sex, age, expanded disability status scale (EDSS) score and prior relapse rate, the ozanimod 0.5 mg group had fewer baseline GdE lesions (0.9) than the other two groups (1.4 in placebo and 1.3 in ozanimod 1.0 mg group), and there were no subjects of African descent in the ozanimod 1.0 mg group.30 As both of these features are known to be associated with worse prognostic outcomes, their underrepresentation in the study suggests a potential bias towards better outcomes for ozanimod.2,31,32
The primary study endpoint of mean cumulative number of GdE lesions on MRI was significantly lower in the ozanimod groups (1.5 in both 0.5 mg and 1.0 mg groups; p<0.0001 for both) compared with placebo (11.1). For the secondary study endpoint of mean number of GdE lesions, 0.3 and 0.2 lesions were reported in the ozanimod 0.5 mg and 1.0 mg groups, respectively versus 3.2 lesions in the placebo group (p<0.0001).30
In the subsequent 2-year, dose-blinded extension of the RADIANCE study (Part A; ClinicalTrials.gov Identifier: NCT01628393),33 participants in the 0.5 mg and 1.0 mg ozanimod groups continued their original dose assignments, while participants in the placebo group were re-randomised to either the 0.5 mg (n=126) or 1.0 mg (n=123) ozanimod groups and followed for 24 months to evaluate the efficacy and safety of the drug. The results of this study confirmed the efficacy findings of the core study with maintenance of low numbers of MRI lesions and annualised relapse rate (ARR) for participants remaining on ozanimod and lower number of MRI lesions and ARR in those who switched from placebo to ozanimod.30,33
Phase III
The safety and efficacy of ozanimod have been evaluated in two phase III studies: SUNBEAM and RADIANCE Part B. RADIANCE Part B (ClinicalTrials.gov Identifier: NCT02047734) was a multicentre, parallel-group randomised, double-blind, controlled study.34 A total of 1,313 patients were randomised to one of three therapies: oral ozanimod 0.5 mg daily, oral ozanimod 1.0 mg daily, or intramuscular IFN beta-1a 30 µg weekly, and were followed for 24 months. The primary study endpoint, the ARR, was lower in the ozanimod groups in comparison with the IFN beta-1a group. ARR at 24 months was 0.22 and 0.17 in ozanimod 0.5 mg/day and 1.0 mg/day groups compared with 0.28 in the IFN beta-1a group (p=0.0167 for ozanimod 0.5 mg and p<0.0001 for ozanimod 1.0 mg versus IFN beta-1a). There were 2.09 and 1.84 new or enlarging T2 lesions on MRI in the ozanimod 0.5 mg/day and 1.0 mg/day groups compared with 3.18 lesions in the IFN beta-1a group, while there were 0.20 and 0.18 GdE lesions in the ozanimod 0.5 mg/day and 1.0 mg/day groups compared with 0.37 in the IFN beta-1a group (p=0.0030 for ozanimod 0.5 mg and p=0.0006 for ozanimod 1.0 mg versus IFN beta-1a, respectively). Notably, although significant differences in efficacy for the two doses of ozanimod compared with IFN beta-1a were seen, all groups had a relatively small number of GdE lesions.34
Similarly, SUNBEAM (ClinicalTrials.gov Identifier: NCT02294058) was a multicentre, randomised, double-blind, double-dummy, parallel-group, active-treatment controlled design, which enrolled 1,346 patients with RRMS.35 Patients were followed for 12 months for the same primary and secondary endpoints as the RADIANCE Part B study. Demographics were similar to those of RADIANCE. Caucasians and Eastern Europeans represented 99% and 93% of the study participants, respectively. The issue of generalisability of these results therefore should be noted. Both the 0.5 mg/day and 1.0 mg/day ozanimod treatment groups showed significantly lower ARR, as well as significantly fewer new or enlarging T2 or GdE lesions on MRI compared with the IFN beta-1a group. ARR at 12 months of treatment was 0.24 and 0.18 in the ozanimod 0.5 mg/day and 1.0 mg/day groups, respectively, compared with 0.35 in the IFN beta-1a group (p<0.0001 and p=0.0013, respectively). There were 2.14 and 1.47 new or enlarging T2 lesions on MRI in the ozanimod 0.5 mg/day and 1.0 mg/day groups, respectively, compared with 2.84 lesions in the IFN beta-1a group (p<0.0001 and p=0.0032, respectively), while there were 0.29 and 0.16 GdE lesions in the ozanimod 0.5 mg/day and 1.0 mg/day groups, respectively, compared with 0.43 in the IFN beta-1a group (p<0.0001 and p=0.0182, respectively).35
Subjects who completed either one of the phase III studies were offered enrolment in an open-label extension trial, DAYBREAK. Results were presented at the 71st AAN Annual Meeting in May 2019 and are not yet published.36 In this study, patients from the two phase III trials were switched to ozanimod 1.0 mg/day and ARR was compared between groups. ARR decreased from 0.184 (95% confidence interval [CI] 0.151–0.224) to 0.131 (95% CI 0.101–0.169) in patients switched from ozanimod 0.5 mg/day (n=756) and from 0.246 (95% CI 0.204–0.297) to 0.126 (95% CI 0.097–0.163) in patients switched from IFN beta-1a (n=741) over the mean duration of 18.5 months of follow-up. Patients who continued on ozanimod 1.0 mg (n=760) also showed a slight reduction in ARR from 0.153 (95% CI 0.125–0.187) to 0.133 (95% CI 0.104–0.172).36 Although these results provide evidence supporting superior efficacy of ozanimod to IFN beta-1a, they must be interpreted with caution, as a negative binomial regression was used to calculate ARR in the parent and open-label extension trials in this interim analysis with adjustments for multiple factors.
Another selective S1PR1 and S1PR5 modulator, siponimod, has been tested in RRMS in phase II trials and in progressive MS in a phase III trial. The phase II studies showed a significant decrease in the number of new or enhancing T2 MRI lesions with siponimod compared with placebo.37,38 In the phase III trial (secondary progressive MS), participants on siponimod had significantly lower confirmed disability progression compared with placebo, with a hazard ratio of 0.79 at 3 months.23 Ozanimod and siponimod have not been compared head-to-head in any trials, but, given these results, their efficacy profiles may be similar.
Secondary outcomes – magnetic resonance imaging and functional metrics
The phase III clinical trials also evaluated changes in whole brain volume, cortical grey matter volume and thalamic volume in addition to changes in functional measures of impairment including the MS functional composite (MSFC), MSFC with low contrast letter acuity (LCLA), symbol digit modalities test (SDMT), and the 54-item multiple sclerosis quality of life (MSQOL-54).34 After adjusting for region, baseline EDSS and baseline value of interest, the authors reported that patients in the ozanimod groups had significantly less brain volume loss: difference in mean percentage change in the ozanimod groups compared with IFN beta-1a was 0.12–0.24 for whole brain volume, 0.61–0.84 for cortical grey matter volume and 0.34–0.59 for thalamic volume. These secondary analyses were performed in patients who had available data, and imputation was used for missing MRI data in 10–12% of participants for MRI measures. Mean scores of MSFC (difference from IFN beta-1a was 0.101, p=0.0246) and MSFC with LCLA (difference from IFN beta-1a was 0.093, p=0.0123) decreased in patients receiving ozanimod 0.5 mg in the RADIANCE Part B cohort; however, these effects were neither observed with ozanimod 1.0 mg nor reproduced in the SUNBEAM trial.34 Of the sub-scores of the MSQOL-54, only the physical health composite scores decreased (decrease of 1.849 points [p=0.0228] in the ozanimod 0.5 mg group in RADIANCE Part B and 1.642 [p=0.0364] in the SUNBEAM trial compared with IFN beta-1a). In SUNBEAM, the SDMT Z score was significantly better in both the ozanimod 0.5 mg (difference versus IFN beta-1a 0.082, p=0.0246) and 1.0 mg groups (difference versus IFN beta-1a 0.111, p=0.0024).35
The significance of these results is unknown; improvements in scores related to functional measures of impairment were seen mostly at lower ozanimod dosages. Notably, the groups were well balanced in terms of disease duration, disability status and baseline number of MRI lesions – the authors also adjusted for region due to overrepresentation of Eastern Europeans and Caucasians in this cohort (98% and 86% of the study population, respectively).
Clinical safety
Phase I
Safety analyses in 68 ozanimod-treated and 24 placebo-treated healthy individuals in a phase I trial demonstrated that the most common treatment-emergent adverse events (TEAE) were contact dermatitis reported in 29 subjects, followed by headaches, somnolence, nausea, dizziness and fatigue – each of these were seen in <10 subjects.17 The rest of the TEAEs were seen in <5% of the subjects and included pruritus, abdominal pain, abnormal dreams, dry mouth, puncture-site haematoma, sinus arrest, oral herpes, cough, nasal congestion, obstructive airway disorder, rhinorrhoea, wheezing, decreased appetite, neck pain. None of the latter were seen in placebo groups, except for abnormal dreams that occurred also in one placebo control subject. One subject in the ozanimod 0.3 mg cohort had bronchoalveolar carcinoma that was not thought to be treatment-related, but rather pre-existing and missed on pre-trial screening.17
A first-dose bradycardia effect was greater in ozanimod-treated subjects in a dose-dependent manner compared with placebo-treated subjects. Cardiac TEAEs including bradycardia with sinus arrest (n=2, duration 2 seconds), intermittent bradycardia (n=1), and second-degree Mobitz type 1 atrioventricular block (n=1, study discontinuation) were reported. The authors of the study found that gradual dose escalation reduced the rate of first-dose cardiac effects after comparing different dosing schemes.17
Phase II
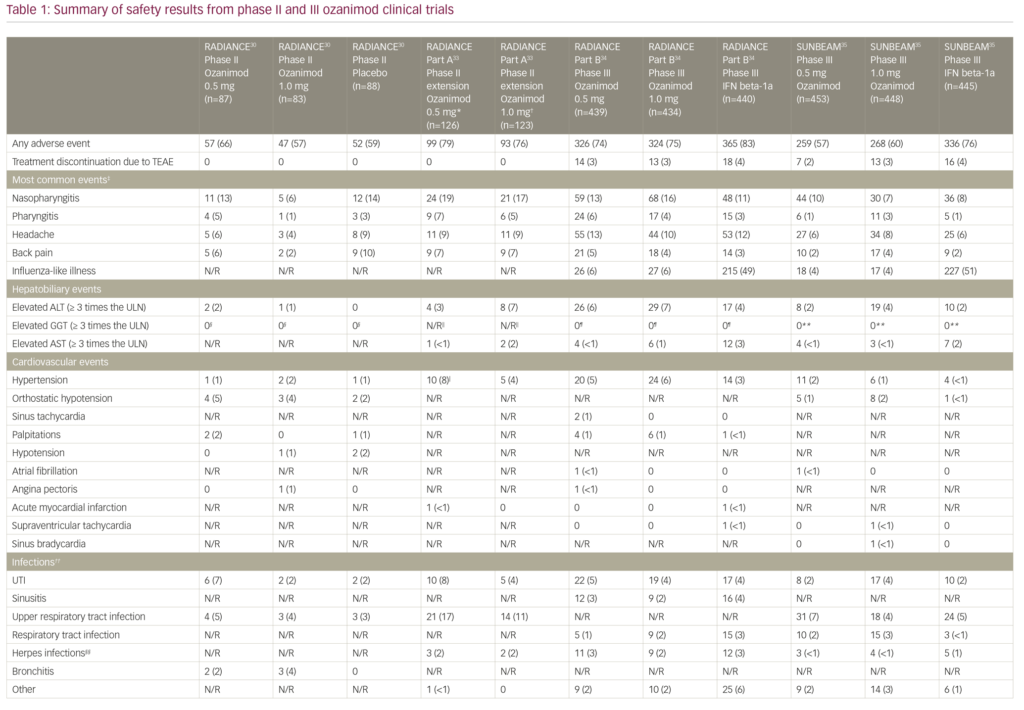
Safety data from the pivotal clinical trials are summarised in Table 1.30,33–35 The rates of adverse events in the core RADIANCE study were comparable between groups, and no subjects discontinued ozanimod due to safety concerns.30 The majority of reported cardiovascular adverse events were orthostatic hypotension and hypertension, while the remaining cardiovascular adverse events were reported in less than 2% of the patients.30 First-dose cardiac effects were monitored with the use of 24-hour Holter monitoring in all groups and showed that the maximum mean change in heart rate was +0.7 beats per minute (bpm) in the placebo group and -1.9 bpm in the combined ozanimod group, while minimum hourly rate did not decrease below 45 bpm in any of the ozanimod-treated subjects.30
The adverse events recorded in the RADIANCE Part A extension trial were similar to those reported in the core study (Table 1).30,33 Liver enzyme abnormalities were among the most common TEAEs. Alanine aminotransferase (ALT) was elevated to at least three times the upper limit of normal (ULN) in four patients in the ozanimod 0.5 mg group and eight patients in the ozanimod 1.0 mg group. Aspartate aminotransferase (AST) was also elevated in three patients: one patient each in the ozanimod 0.5 mg and 1.0 mg groups, both with concomitant ALT elevation, and another patient in the ozanimod 1.0 mg group with isolated AST elevation. Overall, four participants discontinued the extension study due to a >5 times ULN elevation in liver transaminases.33 In terms of cardiac adverse events, asymptomatic bradycardia (44 bpm) was recorded in one patient who was switched from placebo to ozanimod 1.0 mg. There were no reports of atrioventricular block, serious opportunistic infections, clinically significantly pulmonary function test abnormalities, cases of macular oedema, malignancy or treatment-related deaths in either of the groups during the 2-year blinded extension period.33
Phase III
The adverse event profile was similar between the two phase III studies (Table 1): TEAEs were reported in 650/873 of ozanimod-treated patients in RADIANCE Part B and 527/901 of ozanimod-treated patients in SUNBEAM.34,35 Overall, rates of serious adverse events and study discontinuation were low across all three treatment groups (ozanimod 0.5 and 1.0 mg daily, and IFN beta-1a 30 µg weekly). There were no reports of serious opportunistic infections, or second-degree or higher atrioventricular blockage in RADIANCE Part B or SUNBEAM studies.34,35 Only one death occurred which was due to an accidental drowning in a subject in the ozanimod 0.5 mg arm of the RADIANCE Part B study.34 The most common adverse events leading to discontinuation in the phase III studies included elevation of liver enzymes, urticaria, influenza-like illness, back pain and headache in ozanimod treatment groups.34,35
Hypertension was reported in 61/1,774 of ozanimod- versus 18/885 IFN beta-1a-treated patients.34,35 Serious cardiac TEAEs including sinus tachycardia, sinus bradycardia, atrial fibrillation, acute myocardial infarction, supraventricular tachycardia, and angina pectoris were seen in <1% of participants in ozanimod groups.34,35 Both studies used a 7-day dose-escalation regimen for starting participants on ozanimod. With regards to first-dose bradycardia, the authors reported a 1.8 bpm maximum reduction in mean supine heart rate at 5 hours after administration of ozanimod 0.25 mg in the RADIANCE Part B trial.34 Compared with IFN beta-1a, ozanimod-treated patients had a greater incidence of first-dose bradycardia: 9/439 patients in the ozanimod 0.5 mg group and 13/434 patients in the ozanimod 1.0 mg group had a minimum supine heart rate of <50 bpm (none were <40 bpm), while none of the patients in the IFN beta-1a group had a <50 bpm minimum supine heart rate. One patient had dizziness with a heart rate of 47 bpm at 6 hours after administration of the first dose that was treated with subcutaneous atropine. The patient had a history of autonomic dysregulation.34
The cardiac safety profiles reported in the abovementioned studies on ozanimod are similar to those reported for siponimod in the phase III EXPAND trial: hypertension was seen in 115/1,099 of siponimod 2.00 mg/day (titrated from 0.25 mg/day over a week at trial onset) versus 41/546 of placebo-treated patients, thromboembolic events were seen in 33/1,099 versus 15/546, first-dose bradycardia was seen in 48/1,099 versus 14/546, and bradyarrhythmia was seen in 29/1,099 versus 2/5,460. There were no reported cases of atrioventricular block.23 Notably, the rate of first-dose bradycardia was similar in individuals treated with siponimod, those treated with ozanimod 1.00 mg, and patients with RRMS in the phase III trial of fingolimod in 2014, which showed
first-dose bradycardia in 21/370 of patients (15 symptomatic) treated with fingolimod 1.25 mg/day and 5/358 of patients (three symptomatic) treated with fingolimod 0.50 mg/day versus 1/355 patients in the placebo group.10
A large open-label observational study of first-dose cardiac effects in fingolimod-treated patients with RRMS identified atrioventricular block in 62/3,951 and bradycardia in 31/3,951, a rate of first-dose bradycardia much lower than the phase III trials of fingolimod, and lower than in either the siponimod or ozanimod trials.39 Whether this was related to selection bias is unclear, but it is derived from a real-world cohort, and this should be kept in mind when assessing these numbers. It is therefore not clear whether the rate of cardiac adverse events is lower in patients treated with ozanimod in comparison to siponimod or fingolimod. Despite gradual dose-escalation in ozanimod-treated patients, the ozanimod trials demonstrate that first-dose bradycardia is still higher using this agent than in patients treated with IFN beta-1a.
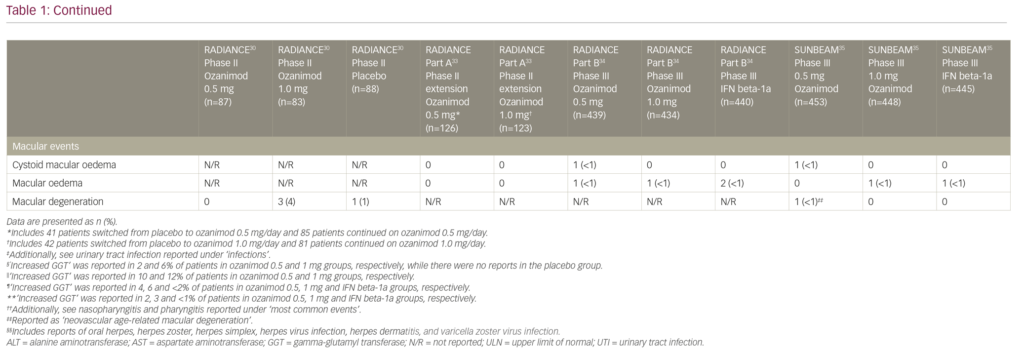
Macular abnormalities including macular oedema (cystoid or not), macular hole, maculopathy and neovascular age-related macular degeneration were reported in 5/1,774 of patients treated with ozanimod and 3/885 of patients treated with IFN beta-1a in phase III trials. All of these events led to treatment discontinuation.34,35 In comparison, macular oedema was seen 2/370 patients in the fingolimod 1.25 mg/day group and 1/358 patients in the 0.50 mg/day group, versus 1/355 patients in the placebo group in the phase III FREEDOMS trial.10 A recently reported long-term safety analysis of fingolimod also supported these findings: over the observation period of up to 14 years 1/4,086 patients had macular oedema that was recorded at year 5.40 However, reports of macular oedema were slightly more common in the phase III siponimod cohort: macular oedema was seen in 18/1,099 patients in the siponimod 2.00 mg/day group versus 1/546 patients in the placebo group.23 Overall, macular oedema remains as a rare but serious adverse effect of S1PR modulators, and neither of the selective agents appears to provide a clear advantage in this regard.
Abnormalities in liver function tests were seen in 8.6% of ozanimod-treated patients in phase III trials. In RADIANCE Part B 4.5% of patients and in SUNBEAM 5.4% of patients in the IFN beta-1a group had elevated ALT (>3 times the ULN). AST and total bilirubin elevations were less common and occurred in <3% of subjects in both the ozanimod and the IFN beta-1a groups.34,35 Overall, abnormalities in the levels of hepatobiliary enzymes including ALT, AST, bilirubin and gamma-glutamyl transferase led to treatment discontinuation in 16/1,771 ozanimod participants and 8/885 IFN beta-1a participants included in the safety analyses in phase III clinical trials.34,35 These results are comparable to trials of siponimod and fingolimod; liver enzyme abnormalities were reported in 12% and 21% of siponimod and fingolimod groups, respectively in the phase III EXPAND and FREEDOMS trials.10,23 Infections were reported in 617/1,099 patients treated with siponimod 2.00 mg/day versus 287/546 patients receiving placebo; 269/370 and 263/358 patients treated with fingolimod 1.25 mg/day and 0.50 mg/day, respectively, versus 255/355 patients receiving placebo; while in 240/892 patients treated with ozanimod 0.50 mg/day; and 238/882 patients treated with ozanimod 1.00 mg/day versus 221/885 IFN beta-1a-treated patients. Of note, convulsions, which were not reported in ozanimod trials, were seen in 2% of the patients in the siponimod trial and 1% of the patients in the fingolimod trial compared with <1% of the placebo groups.23
Discussion
Above, we have reviewed studies of the safety and efficacy of ozanimod in MS, an oral S1PR modulator with improved selectivity for the S1PR1 and S1PR5 subtypes. It appears to be well tolerated in individuals with MS up to the maximum dose of 3 mg/day in phase II and III clinical trials, and to have superior efficacy to IFN beta-1a at both doses studied. While it was created in order to avoid key adverse effects reported in association with the FDA-approved S1PR modulator, fingolimod, it is not entirely clear that it achieves this goal: cardiac first-dose effects including negative chronotropy have been reported in published studies. It remains to be seen whether this therapy has the hoped-for safety advantages over fingolimod and the other approved agent, siponimod. Nonetheless, trials outlined in this review suggest that this therapy may provide an effective additional medication to add to the growing armamentarium of therapies for MS. Future studies will establish the long-term benefits of this therapy as well as its effects on disease progression.