Multiple sclerosis (MS) is a chronic neuro-inflammatory condition estimated to affect over two million people worldwide. Several subtypes of MS have been described, and they are defined by their clinical phenotypes.1 The majority of patients initially exhibit relapsing remitting MS (RRMS), which is characterized by acute episodes of overt inflammation associated with abrupt clinical decline, most often accompanied by new magnetic resonance imaging (MRI) lesions, the hallmark of the disease. Patients with RRMS notably experience no neurological worsening outside of abrupt clinical attacks. After one to two decades, a minority of patients with RRMS subsequently develop gradual neurological worsening aside from that caused by clinical relapses, generally accompanied by a decrease in MRI lesion accrual, and an increase in brain atrophy rate. This phase, which is characterized by less overt inflammation and more neurodegeneration, is termed secondary progressive MS (SPMS). Importantly, there is overlap between the subtypes, and patients with SPMS may still experience overt inflammation in the form of new MRI lesions or superimposed relapses, albeit typically at a reduced rate than in those with RRMS. This is referred to as “active” SPMS.1 While there are more than 15 US Food and Drug Administration (FDA)-approved disease-modifying therapies for RRMS, there has been a relative dearth of treatments for progressive forms such as SPMS.
Siponimod (Mayzent®, Novartis, Basel, Switzerland) is a second-generation oral sphingosine- 1-phosphate (S1P) receptor modulator, approved by the FDA in 2019 for the treatment of relapsing forms of MS, including active SPMS.2 In this article, we review the clinical trial data regarding the use of this medication and the implications for use in patients with SPMS.
Mechanism of action—sphingosine-1-phoshate receptor modulation
The S1P receptor family consists of five subtypes (S1P1, S1P2, S1P3, S1P4, and S1P5). These cell-surface receptors are ubiquitous and serve important functions in the central nervous, immune, cardiovascular, and respiratory systems.3 There are three FDA-approved agents for MS that target the S1P receptor family—fingolimod (Gilenya®, Novartis, Basel, Switzerland),4 siponimod (Mayzent),2 and ozanimod (Zeposia®, Bristol Myers Squibb, New York, NY, USA).5 Efficacy against MS is thought to be primarily mediated by S1P1 binding on lymphocytes, leading to receptor internalization from the cell-surface and functional antagonism. This receptor is necessary for lymphocyte egress from lymphoid tissue, and its antagonism leads to lymphocyte sequestration in lymph tissues, thereby preventing central nervous system (CNS) invasion.3
As S1P receptors are also present in various cells of the CNS (neurons, oligodendrocytes, astrocytes, and microglia),3 it is postulated that S1P modulators might also exhibit neuroprotective effects. In a mouse experimental autoimmune encephalitis model of MS, S1P receptor modulators have helped to promote remyelination and prevent neurodegeneration.6,7 While the non-selective S1P receptor modulator fingolimod has been used in the treatment of RRMS for many years,4 siponimod and ozanimod are new agents that selectively target S1P1 and S1P5 receptors. As some of the adverse effects of S1P modulators are mediated by S1P2–4 (cardiac effects, for example, are thought to be mediated through S1P1 and S1P3 in cardiac myocytes), it is hypothesized that this selectivity can reduce adverse effects while preserving the effects on lymphocyte sequestration and the CNS.2,5 However, despite increased selectivity, the risk profile of siponimod and ozanimod is similar to that of fingolimod.2,4,5 Compared with fingolimod, siponimod has a short half-life, leading to washout in 7–10 days and reversal of effects within several weeks.2
Clinical efficacy
Phase II
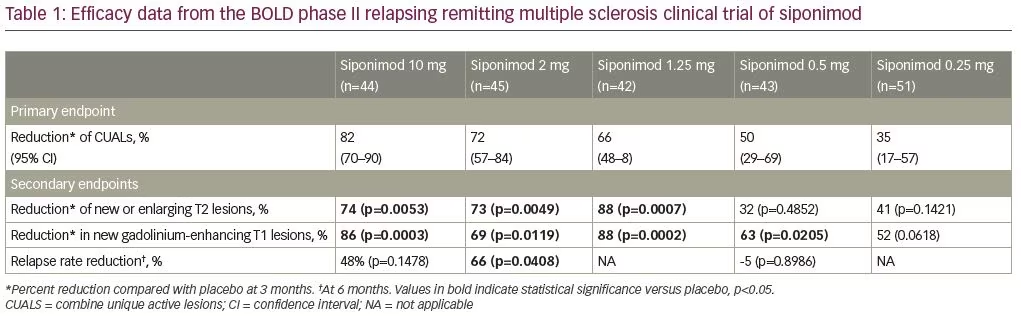
A dose-finding, placebo-controlled, double-blind phase II study of siponimod was performed in patients with RRMS (BOLD, ClinicalTrials.gov identifier: NCT00879658).8 The study recruited two patient cohorts, separated by a 3-month interim analysis. Cohort 1 (n=188) consisted of patients with RRMS aged 18–55 years, randomized in a 1:1:1:1 ratio to receive daily siponimod at 10 mg, 2 mg, 0.5 mg, or placebo for 6 months. Patients in cohort 2 (n=109) were randomly assigned in a 4:4:1 ratio to siponimod 1.25 mg, siponimod 0.25 mg, or placebo for 3 months. The study demonstrated a dose–response relation across all doses of siponimod tested. Doses of 10 mg, 2 mg, 1.25 mg, and 0.5 mg all significantly reduced the number of new brain lesions at 3 months compared with placebo. Additionally, the study reported a decrease in annualized relapse rate (ARR) of 65% with the 2 mg daily dose, compared with placebo. See Table 1 for a summary of clinical trial results.
Phase III
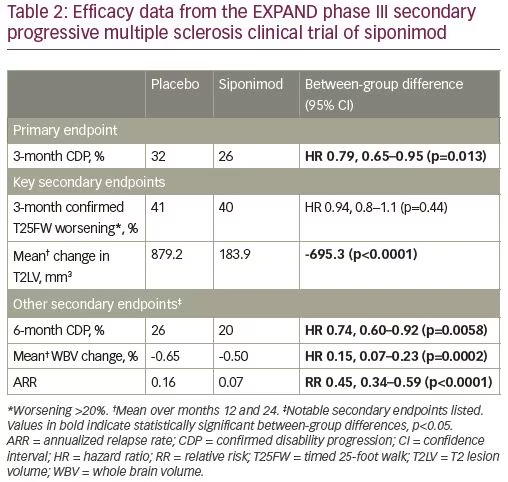
In 2012, a large, randomized, double-blind, placebo-controlled, event-driven phase III trial of siponimod (EXPAND, NCT01665144) was started for patients with SPMS.9 Eligible patients were aged 18–60 years, had a diagnosis of SPMS, a baseline expanded disability status scale (EDSS) of 3.0–6.5 (6.5 is the highest possible score for an ambulatory patient), progressive EDSS worsening over 2 years prior to enrollment, and an absence of relapses in the 3 months prior to randomization.9 Patients who had other substantial immunological, cardiac, or pulmonary comorbidities were excluded from the study. Those with macular edema or uncontrolled diabetes were also excluded due to concern for treatment-related adverse events. A total of 1,651 patients were randomized in a 2:1 ratio to once daily oral siponimod 2 mg or placebo. The medication was titrated from 0.25 mg to 2 mg over 6 days to limit cardiovascular adverse effects (see section on clinical safety). The primary endpoint was defined as time to 3-month confirmed disability progression (CDP). This was defined as a 1-point increase in EDSS score from a baseline range of 3.0–5.0, or a 0.5-point increase from a baseline score of 5.5–6.5. The major secondary endpoints included overall accrual of T2 lesion volume from baseline, and time to 3-month confirmed 20% slowing of walking speed as measured using the timed-25-foot walk test.
The study was completed after a total of 374 3-month CDP events occurred. The median duration on the study was 21 months and the median exposure to siponimod or placebo was 18 months. Overall, 26% of patients on siponimod and 32% of patients on placebo had 3-month CDP, a statistically significant, but only moderate, clinical effect consisting of a 21% risk reduction.9 A planned subgroup analysis demonstrated that patients exhibiting superimposed relapses in the 2 years before enrollment (i.e., active SPMS) exhibited a statistically significant 33% reduction in 3-month CDP, whereas the reduction in patients without a recent relapse (i.e., “inactive” SPMS) was 13% and was not statistically significant. Siponimod did not influence walking speed. Other secondary endpoints demonstrated an 80% reduction in number of new T2 lesions, a 55% reduction in ARR, and a 23% reduction in whole brain volume loss over 2 years.9 See Table 2 for a summary of clinical trial results.
FDA approval and indication
As the EXPAND study demonstrated a statistically significant effect on CDP only in patients with superimposed relapses over the prior 2 years, siponimod is approved by the FDA for the treatment of active SPMS. Although siponimod demonstrated a modest benefit in patients with SPMS without prior relapses, the FDA did not approve siponimod for inactive SPMS.1,2 Based on results from the BOLD, EXPAND, and prior fingolimod studies, siponimod is also approved for other relapsing forms of MS, such as clinically isolated syndrome and RRMS.4
Initiation protocol
Siponimod is metabolized by the cytochrome P450 system, primarily CYP2C9 and CYP3A4. As genetic variations may impact metabolism of the drug, genotype testing is required to determine optimal dose of the medication.2 The majority of patients carry CYP2C9 genotypes *1/*1, *1/*2, or *2/*2, which necessitate a maintenance dose of 2 mg daily starting on day 6 after an up-titration of treatment (0.25 mg on days 1 and 2, 0.5 mg on day 3, 0.75 mg on day 4, and 1.25 mg on day 5). In patients with CYP2C9 *1/*3 or *2/*3 genotypes, which lead to slower drug metabolism, the recommended maintenance dose is lower—1 mg daily starting on day 5 of treatment after 4 days of up-titration.2 Siponimod is contraindicated in patients who are homozygous for CYP2C9*3, which constitutes only about 0.3–0.4% of Caucasians, and an even lower rate in other ethnic populations.
Unlike with fingolimod, a first-dose observation (FDO) is not required unless a patient has a cardiac comorbidity (see clinical safety below).2 While siponimod holds some benefits over fingolimod with regards to initiation and monitoring requirements, step-edits and insurance prior authorizations may currently limit the use of siponimod in RRMS and clinically isolated syndrome.
Clinical safety and tolerability
Siponimod’s safety and tolerability profile is similar to that of other members in its class and is primarily dependent on its effects on S1P receptors (see Table 2 for summary). The most common adverse reactions in EXPAND were headache (15% on drug, 14% on placebo), hypertension (13% on drug, 9% on placebo), and transaminitis (11% on drug, 3% on placebo).9 As such, physicians should exercise caution when using siponimod in patients with hypertension or prior liver disease. Patients in the EXPAND trial who developed transaminitis above three times the upper limit of normal discontinued the use of siponimod.9 Therefore, it is recommended that blood work be monitored regularly and siponimod be discontinued if substantial transaminitis is noted.
Initiation of siponimod is associated with a mild and asymptomatic temporary bradycardia and atrioventricular conduction delay. This is mediated by effects on S1P1 receptors in cardiac myocytes. Therefore, an electrocardiogram is required in all patients prior to starting the medication. While fingolimod requires FDO,4 the slow up-titration protocol of siponimod over several days (dependent on CYP2C9 genotype, see above) limits the cardiac risk. Therefore, FDO is not required for most patients when administering siponimod. While the lack of FDO requirement is regarded as an advantage, the up-titration needs to be repeated starting at day 1 if a patient misses just one of the titration doses.2 Given the short half-life, if four or more consecutive maintenance doses are not taken, the up-titration protocol needs to be re-initiated. This is potentially less advantageous than fingolimod, where patients are required to repeat the FDO only if 14 consecutive days are missed. In patients with prior cardiac comorbidities such as mild sinus bradycardia, first or second-degree atrioventricular block, or myocardial infarction >6 months prior to initiation, a cardiology consultation and FDO is recommended before starting siponimod. A history of cardiac arrest, myocardial infarction within the previous 6 months, or uncontrolled hypertension serve as relative contraindications, and alternatives to siponimod should be considered. In patients taking medications that may cause bradycardia, such as beta-blockers or calcium channel blockers, or medications that can prolong QT, it is recommended to temporarily withhold such treatments during initiation of siponimod to avoid compounded adverse effects. This, however, should be done with the consultation of a cardiologist, and the potential benefit of siponimod should be weighed against the risks of holding these medications.2
Siponimod is metabolized by the cytochrome P450 system; as such, there is increased potential for complex drug interactions above what is typically considered with fingolimod. Use of medications that cause both moderate CYP2C9 and moderate or strong CYP3A4 inhibition (e.g. fluconazole) is not recommended due to a resultant rise in siponimod drug level. Similarly, use of medications that cause both moderate CYP2C9 and strong CYP3A4 induction (e.g., rifampin, tegretol) is not recommended, as this leads to decreased siponimod levels. Finally, it is important to note that moderate or strong CYP3A4 inducers are not recommended in patients with patients who have CYP2C9*1/*3 or *2/*3 genotypes; this includes modafinil, which is a mainstay treatment of MS-related fatigue.2
Siponimod causes a dose-dependent decrease in peripheral lymphocyte counts to 20–30% of baseline values,2,9 consistent with its known mechanism of action. This leads to an increased risk for typical and opportunistic infections. In the phase III EXPAND trial, the rate of infections in total was similar in both drug and placebo groups (49% versus 49.1%). However, herpes/zoster, bronchitis, sinusitis, upper respiratory infections, and fungal skin infections were more common in the siponimod group. Serious infections were mildly increased (2.9%, versus 2.5% on placebo). Cryptococcal meningitis, although rare, has been reported with siponimod, and close monitoring is indicated.2 Zoster reactivation leading to meningitis has been reported, as with fingolimod, and patients require testing for immunoglobulin G (IgG) antibodies to varicella-zoster (VZV) prior to starting siponimod.2 Patients without VZV IgG seropositivity undergo vaccination 1 month prior to initiation of siponimod. While progressive multifocal leukoencephalopathy (PML) has not yet been reported with siponimod, more than 20 cases have been reported with fingolimod, and incidence appears to be higher with increased patient age.4 As the mechanism of action is similar, it seems reasonable to expect rare cases of siponimod-related PML as use increases over time. As siponimod is indicated for, and is often used in, an older population (i.e., those with active SPMS), vigilance for PML is highly recommended. More post-marketing experience with siponimod will be necessary to conclusively determine the risk of PML.
In order to mitigate the risk of infections, patients initiating siponimod require evaluation with a complete blood count, and those with active infection should not begin siponimod until resolution. If a serious infection occurs during treatment, the drug should be held until resolution of the infection. Siponimod has a short half-life, so washout occurs within 7 days. Residual effects may persist for 3–4 weeks after cessation.2
As with fingolimod, evaluation for macular edema is indicated with siponimod. Macular edema was reported in 1.8% of patients on siponimod compared with only 0.2% on placebo.9 Diabetes mellitus or prior history of uveitis substantially increases the risk of macular edema with siponimod, and a careful consideration of the risks and benefits of siponimod should be undertaken in patients with such conditions. As patients with uncontrolled diabetes were excluded from the EXPAND trial,9 the safety of siponimod in this population is unclear. Ophthalmological evaluation prior to siponimod initiation is mandatory for all patients. Surveillance ophthalmological examinations may detect early mild macular edema even in asymptomatic patients, so it is recommended for patients to undergo repeat ophthalmological examinations during peak risk, 3–4 months after starting siponimod.2
Siponimod is potentially teratogenic, and adequate contraception during treatment is required in women of childbearing age. Since the half-life of siponimod is short and the drug is washed-out after 10 days,2 siponimod may offer a family-planning advantage over fingolimod, which should be discontinued at least 2 months prior to conception.
Conclusion
Siponimod is a novel selective S1P receptor modulator that has demonstrated substantial efficacy in the treatment of RRMS, with modest efficacy in delaying disability worsening in SPMS, mostly in active SPMS. The benefit of siponimod was statistically significant only in those with active SPMS, with a modest and non-significant effect in patients without recent relapses. Hence, siponimod is approved by the FDA only for use in active SPMS. The number of FDA-approved S1P receptor modulators continues to rise,2,4,5 and several other medications have been granted FDA approval for the treatment of active SPMS based on efficacy in RRMS. However, siponimod remains the only modern medication to have demonstrated efficacy in a trial of SPMS patients (mitoxantrone had previously obtained FDA approval for SPMS in 2000, but has since fallen out of favor due to poor safety and tolerability).
Siponimod is a potent anti-inflammatory agent with a substantial effect on relapse rate. Based on preclinical studies, an additional neuro-protective role is possible,6,7 but this has not been elucidated in clinical studies. A neuro-protective effect might mediate efficacy in progressive MS. However, as much of the effect in SPMS was seen in patients with active SPMS, it is possible that its efficacy in SPMS may still be mediated by relapse reduction alone. This is reminiscent of results from studies on ocrelizumab, which demonstrated potent relapse reduction, but only modest effects on CDP in primary progressive MS.10 The idea that potent reduction in relapse rate can prevent disability in progressive MS is consistent with evidence that relapses may mediate disability worsening in early progressive MS.11 As such, the ideal patient for siponimod may be one with early SPMS, whose disease is likely to be active, or one who is transitioning from RRMS to SPMS. A recent subgroup analysis of the EXPAND study demonstrated a potential cognitive benefit of siponimod, with a higher proportion of patients in the siponimod group having cognitive improvement at the conclusion of the study.12 This is of importance, as cognitive function is a common cause of morbidity in patients with MS in general and SPMS specifically. It is unclear whether this effect is mediated by relapse reduction alone or by another neuro-protective mechanism, and further research is necessary.