Neuromyelitis optica spectrum disorder (NMOSD) refers to a family of inflammatory central nervous system (CNS) diseases in which patients accrue disability through severe episodes of demyelination with typical manifestations including involvement of visual pathways (e.g., optic neuritis) and spinal cord (longitudinally extensive myelitis). Other common symptoms, including intractable hiccups, nausea and vomiting are also well recognized. The majority of patients will have detectable aquaporin-4 immunoglobulin-G (AQP4-IgG) antibodies.1,2 A significant subset of patients who are AQP4-IgG-seronegative have myelin oligodendrocyte glycoprotein (MOG)-directed antibodies, now recognized as a distinct syndrome. Diagnostic criteria for NMOSD are more rigorous for those who are AQP4-IgG-seronegative.3 In contrast to multiple sclerosis (MS), patients with NMOSD rarely demonstrate a progressive course of disability accumulation, but relapses tend to be more severe. Thus, relapse prevention is of the utmost importance in NMOSD.
While treatment of acute NMOSD relapses mirrors that for MS (high-dose corticosteroids and plasma exchange for severe relapses),4,5 many therapies used for prevention of MS attacks have been either ineffective6 or even harmful7–10 in NMOSD. The previous lack of prospective randomized trials to study immunotherapies aimed at preventing new relapses in NMOSD meant that treatment was guided by observational data and influenced by medication availability and cost, medical comorbidities, and patient and physician preferences. Commonly used medications have included rituximab,11–13 mycophenolate mofetil, azathioprine,14 methotrexate, concomitant oral prednisone, and less-frequently, tocilizumab (anti-interleukin [IL]-6-receptor antibody) and mitoxantrone. Novel preventative therapies aimed at relapse reduction (to prevent disability accrual) have recently emerged and target various pathways in the pathophysiology of NMOSD,15–17 briefly summarized below.
Pathophysiology of neuromyelitis optica spectrum disorder
As noted, most patients with NMOSD have detectable IgG1 autoantibodies targeting the AQP4 channel present on astrocyte endfeet. These AQP4-IgG antibodies are secreted by plasmablasts (differentiated B cells), which are thought to develop, in part, through interaction with helper T cells (the Th-17 subset being most implicated), potentially based on recognition of AQP4 peptides. IL-6 is implicated in both the differentiation of naïve T cells into pro-inflammatory Th-17 cells and in the differentiation of B cells into AQP4-IgG-producing plasmablasts. Binding of AQP4-IgG to astrocytes, in turn, is thought to induce a complement-mediated cascade and produces an inflammatory milieu resulting in astrocyte destruction and neuronal injury, leading to the often-severe clinical picture of NMOSD. Herein we focus our discussion on three immunotherapies with positive phase III randomized trials for relapse reduction in NMOSD and discuss how these targeted therapeutics fit into NMOSD clinical practice.
Novel treatments for neuromyelitis optica spectrum disorder
Eculizumab
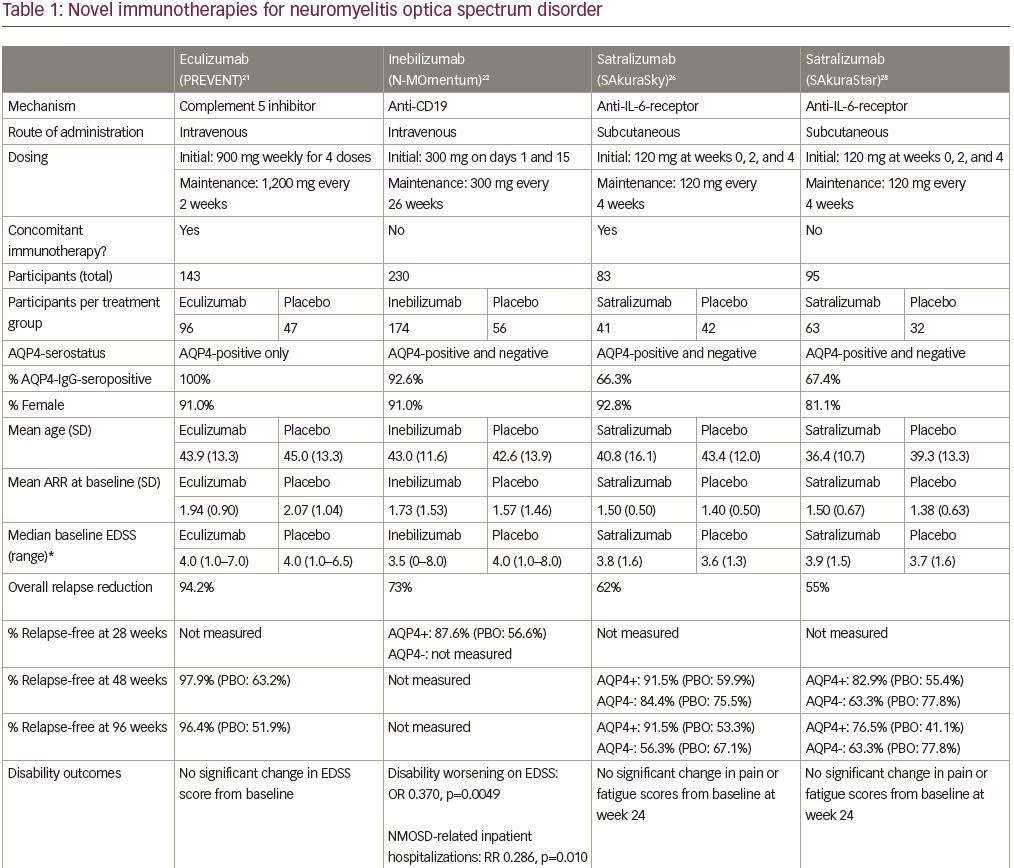
Eculizumab is a humanized monoclonal antibody that inhibits the terminal complement protein (C5), preventing its cleavage into its subunits C5a and C5b,18 which are pro-inflammatory and responsible for formation of the membrane attack complex.19,20 The efficacy and safety of intravenous eculizumab was studied in the multicenter phase III, randomized, double-blind, placebo-controlled, time-to-event Prevention of Relapses in Neuromyelitis Optica (PREVENT) trial in adult patients with AQP4-IgG-positive NMOSD (Table 1; also for dosing and frequency details).21 The primary endpoint of this trial was time to first relapse adjudicated by an independent committee, and six hierarchically ordered secondary endpoints included the adjudicated annualized relapse rate (ARR) and change from baseline on various disability (e.g. Expanded Disability Status Scale [EDSS] score) and quality of life scales. Key exclusion criteria included recent treatment with mitoxantrone (≤3 months), rituximab (≤3 months) or intravenous immune globulin (≤3 weeks), or prednisone doses of >20 mg per day. A cohort of 143 participants with active NMOSD were randomized (eculizumab n=96; placebo n=47) (Table 1) and many (32%) had previously received rituximab. Furthermore, most patients (76% overall) were continued on a stable immunotherapy regimen (most commonly glucocorticoids, mycophenolate mofetil or azathioprine).
Three of the 96 patients (3%) receiving eculizumab, and 20 of 47 (43%) receiving placebo, met the primary endpoint of adjudicated relapse, with a hazard ratio (HR) of 0.06 (95% confidence interval [CI] 0.02–0.20; p<0.001). Patients treated with eculizumab had a significantly lower adjudicated ARR (0.02) when compared with the placebo group (0.35) (p<0.001). Most of the relapses were classified as myelitis. To determine the role of concomitant immunosuppressive therapy, subgroup analysis was performed showing that none of the 21 patients receiving eculizumab alone, and seven of the 13 patients (54%) receiving placebo alone, experienced adjudicated relapses. There was no significant difference between groups in the change in neurologic disability (EDSS). Note that the trial was terminated earlier than planned due to desire to limit harm.
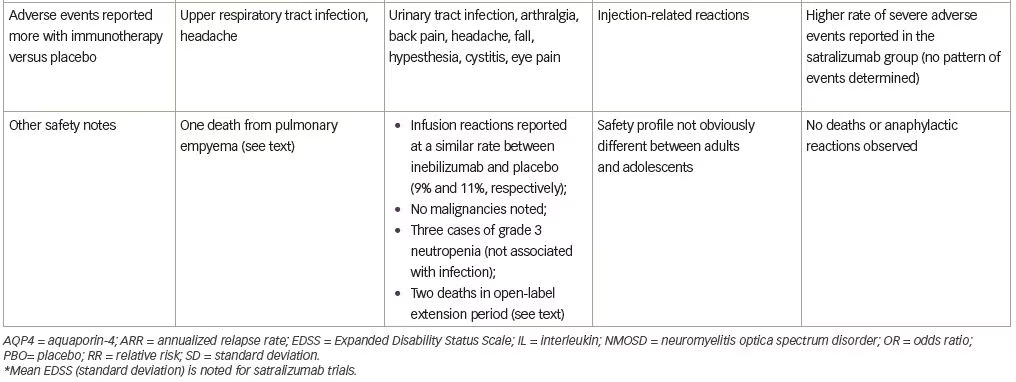
With respect to safety, all patients were vaccinated against Neisseria meningitidis due to the known risk of infection with encapsulated organisms while on complement inhibitor therapy and no patients in the trial developed a meningococcal infection. Upper respiratory tract infection and headache were reported more commonly in the eculizumab group. One patient receiving eculizumab and azathioprine died of a pulmonary empyema.
Overall, eculizumab significantly reduced the risk of relapse in patients with AQP4-IgG-positive NMOSD when compared with placebo. The efficacy and safety of this therapy in the AQP4-IgG-seronegative population and those with more recent exposure to excluded immune therapies remains unknown. Clinicians must remain cognizant that most (76%) participants in the PREVENT trial received concomitant immunosuppressive therapy and only a small subgroup analysis could confirm efficacy of eculizumab in those on monotherapy. In part due to limited follow-up and trial design, eculizumab did not demonstrate a benefit on average disability score change or quality of life outcomes and this should inform how providers counsel patients.
Inebilizumab
Inebilizumab is a humanized, affinity-optimized, afucosylated IgG1 kappa monoclonal antibody binding to the CD19 B-cell surface antigen,22 which depletes a broader range of cells exclusively in the B lymphocyte lineage when compared with anti-CD20 antibodies. The safety and efficacy of intravenous inebilizumab was examined in the N-MOmentum multicenter, double-blind, randomized placebo-controlled phase II/III trial with an open-label extension period (Table 1).22 Adults with active NMOSD (including AQP4-IgG-seronegative participants) were enrolled with exclusions for several medical comorbidities (e.g. prior cancer, diabetes with glycosylated hemoglobin >8%, uncontrolled hypertension). Due to concern for an increase in acute NMOSD attacks following B-cell depletion with rituximab,23 all participants were given prednisone 20 mg per day between days 1 and 14 of therapy with taper. Otherwise, no background immunosuppressant therapy was allowed. The primary outcome was time to the onset of an adjudicated NMOSD attack. Secondary outcomes included percentage of participants demonstrating change from baseline in the EDSS score and low-contrast visual acuity score, cumulative number of active magnetic resonance image (MRI) lesions, and number of NMOSD-related inpatient hospitalizations. Of the 230 participants enrolled (Table 1), only 17 were AQP4-IgG-seronegative (seven of whom tested positive for anti-MOG antibodies).22 While underpowered for formal subgroup analysis, the observation that there were three relapses in 13 patients who were seronegative randomized to the active treatment arm, and no relapses in the other four patients who were seronegative randomized to placebo does not support efficacy in this subgroup.
Overall, treatment with inebilizumab significantly increased the time to first NMOSD attack. Only 21 (12%) of the 174 patients receiving inebilizumab, compared with 22 (39%) of the 56 participants receiving placebo, had an attack in the 6.5-month study period (HR 0.272 [95% CI 0.150–0.496]; p<0.0001) with a number needed to treat of 3.73 (95% CI 3.06–5.66). Fewer inebilizumab-treated participants demonstrated worsening on the EDSS from baseline compared with those treated with placebo (odds ratio [OR] 0.370 [95% CI 0.185–0.739]; p=0.0049), with no difference between groups on the low-contrast visual acuity score over time. Inebilizumab-treated participants had fewer cumulative number of active MRI lesions (relative risk [RR] 0.566 [95% CI 0.387–0.828]; p=0.0034) and had fewer NMOSD-related inpatient hospitalizations (RR 0.286 [95% CI 0.111–0.741]; p=0.010). Overall, significant B-cell depletion occurred in the inebilizumab-treated group, an effect that was durable throughout the randomized controlled period.
Several adverse events were reported more frequently with inebilizumab (Table 1), but interestingly, infusion-related reactions occurred at a similar frequency in both groups (perhaps due to administration of prednisone during the infusion period). Two deaths were reported in the open-label period, both occurring 9 days after a dose of inebilizumab (300 mg). The first death occurred in a participant originally randomized to the placebo arm and was attributed to respiratory insufficiency after a recent NMOSD attack. The second was a participant originally randomized to the inebilizumab group, who died as a result of a CNS process of unclear etiology. The differential diagnosis included acute disseminated encephalomyelitis, atypical NMOSD attack, or progressive multifocal leukoencephalopathy (PML). One of three John Cunningham virus cerebrospinal fluid tests performed on this participant was positive, but two were negative (defined as <10 copies/mL), and MRI findings were not deemed specific to PML. No autopsy data are available for these participants.
In summary, inebilizumab significantly reduced the risk of NMOSD attack and conferred favorable outcomes (less radiographic activity and reduction in disability progression). Efficacy could not be confirmed in the AQP4-IgG-seronegative subgroup and the stringent medical exclusions may also limit the generalizability of these findings. N-MOmentum was also stopped early (randomized phase of 6.5 months), which represents a relative weakness of the data. However, the familiarity that many neurologists already have with B-cell-depleting therapy may translate into popular acceptance of inebilizumab, particularly when considering the lower rate of infusion-related reactions reported in N-MOmentum compared with those reported in trials for the anti-CD20 therapies rituximab24 and ocrelizumab.25
Satralizumab
Satralizumab is a humanized monoclonal antibody that binds both membrane-bound and soluble IL-6 receptors to block the IL-6 signaling pathways.26 Satralizumab uniquely dissociates from the antigen in a
pH-dependent manner to be released into the bloodstream and bind the antigen again (recycling mechanism), which in turn prolongs the elimination half-life of the drug in plasma.26 The efficacy and safety of subcutaneous satralizumab added to stable immunosuppressant treatment was tested in the SAkuraSky study, a phase III randomized, double-blind, placebo-controlled, parallel-assignment trial with an open-label extension period (Table 1).26 Patients (including adolescents aged 12–17 years), with NMOSD (both seropositive and seronegative), were included. Randomization was stratified by baseline ARR (1 versus >1) and geographic region (Asia versus Europe or other). The primary outcome was the proportion of first protocol-defined relapse and relevant secondary outcomes included change from baseline to week 24 on the visual-analogue scale score for pain and the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) score. A total of 83 participants were enrolled (Table 1), and note that a substantial percentage of patients in this trial resided in Asia (39% in the satralizumab group and 43% in the placebo group).
Satralizumab added to baseline immunosuppressant therapy lowered the risk of NMOSD relapse when compared with adding placebo, as only eight participants (20%) receiving satralizumab had a protocol-defined relapse compared with 18 participants (43%) receiving placebo (HR 0.38; 95% CI 0.16–0.88; adjusted p=0.02) (median double-blind treatment duration of 107.4 weeks for satralizumab and 32.5 weeks for placebo). A higher percentage of participants receiving satralizumab remained relapse-free compared with those receiving placebo both at 48 and 96 weeks (89% versus 66% and 78% versus 59%, respectively). There was no significant between-group difference in the change on the pain score from baseline to week 24. Safety was similar between groups (and between adults and adolescents) except that injection-related reactions were more common in the satralizumab group. Other frequently-reported adverse events with satralizumab were nasopharyngitis (24.4%), upper respiratory tract infections (24.4%), and headache (24.4%).
While satralizumab add-on demonstrated longer time to relapse than placebo, it did not demonstrate significant effects on pain or fatigue measures (contrary to the beneficial effect on neuropathic pain and fatigue found for the IL-6 receptor antagonist tocilizumab).27 Inclusion of adolescents with NMOSD is a distinct advantage of this trial, though the relatively small numbers preclude formal conclusions about efficacy and safety in this younger age group.
Satralizumab was also tested in a similarly designed phase III double-blind placebo-controlled, parallel-assignment clinical study in adults with NMOSD as monotherapy (SAkuraStar trial) (Table 1).28 Satralizumab alone reduced the risk of protocol-defined relapse by 55% when compared with placebo (HR 0.45; 95% CI 0.23–0.89; adjusted p=0.018) (Table 1) with no significant effect of satralizumab treatment on pain or fatigue scores. The most common adverse events encountered with satralizumab were upper respiratory tract infections and urinary tract infections. While both SAkuraSky and SAkuraStar trials were underpowered to formally assess efficacy in the participants who were AQP4-IgG-seronegative, neither study identified a beneficial signal in this subgroup.
Discussion
NMOSD relapse can confer significant acute morbidity and sustained disability, making relapse prevention paramount. Selection of the best preventative therapeutic for an individual patient will warrant careful consideration of several factors. Firstly, AQP4-IgG serostatus will dictate eligibility for eculizumab therapy and should influence the way providers counsel their patients about the potential efficacy of other therapies (inconclusive subgroup analyses due to small numbers). This should also motivate researchers to launch prospective clinical trials for patients with anti-MOG antibodies, and to routinely test for these antibodies in future NMOSD trials to better understand efficacy of immune therapies for these patients, who likely comprise a significant proportion of the AQP4-IgG-seronegative NMOSD population.
The lack of head-to-head trials makes treatment selection challenging but some key differences across trials should be considered by providers. Participants across trials were, overall, similar with respect to age, sex, and baseline disability, though the satralizumab trials included the highest percentages of Asian participants, and their add-on trial included adolescent patients. Black or African American patients were included in the studies though may have been underrepresented and their enhanced recruitment could be a goal for future studies. A large proportion of participants in the eculizumab trial were previously treated with rituximab, suggesting eculizumab as an attractive option for those patients with breakthrough disease on anti-CD20 therapy, though the exact timing for transitioning between the two is not fully defined. The inebilizumab trial demonstrated reduction of disability accumulation, and for providers already familiar with anti-CD20 therapy, inebilizumab may be a comfortable choice. Satralizumab, as a self-administered medication, may be appealing to patients. Though no unexpected safety signals emerged from any of the trials, medical comorbidities will also influence treatment choice (e.g. the inebilizumab trial excluded those with significant cardiovascular disease). Long-term safety data is needed for all these agents, but reassuringly, extended use of eculizumab for paroxysmal nocturnal hemoglobinuria29 and myasthenia gravis30 has not revealed surprising safety concerns. Lastly, given the varied study designs employed across trials, it will be of interest to further study the safety and efficacy of these new therapies as monotherapy or in combination with background immune suppression.
For patients with stable NMOSD on non-FDA-approved therapies, coverage by insurance companies may influence the ability to remain on such treatment. Higher costs of particular therapies, which may also vary substantially across jurisdictions, availability and cost of infusion services, and the ability of a patient to adhere to the frequency of therapy (eculizumab and satralizumab are administered more frequently than inebilizumab), pre-treatment requirements (immunizations for eculizumab and steroids for inebilizumab), and monitoring, are all relevant considerations that may influence treatment choice. Importantly, patients will likely have their own preferences in relation to all of the above-mentioned factors. Future trials should address the question of whether lifelong therapy is needed for NMOSD or whether disease-modifying therapy can ever safely be discontinued,31 particularly if these medications can induce a stable state of immune tolerance.
Conclusion
The emergence of three targeted immunotherapies proven to reduce relapse risk in NMOSD has decidedly transformed the landscape of treatment for this rare and severe CNS neuroinflammatory disorder. Further investigation is needed to demonstrate the long-term safety and efficacy of these agents (for relapse and disability prevention) in patients with both AQP4-IgG-seropositive and seronegative NMOSD, and in more diverse and comorbid patients with NMOSD. Despite the uncertainties and challenges that remain, these therapeutic breakthroughs should serve a source of hope for both patients and providers.