Molybdenum cofactor deficiency (MoCD) is an ultra-rare genetic disease that remained untreatable until the recent introduction of cyclic pyranopterin monophosphate (cPMP) substitution. This article aims to comprehensively review the rationale behind cPMP substitution, its effectiveness in clinical scenarios, the development of current treatment protocols, and remaining unmet needs. The clinical manifestations of MoCD, its pathology, current concepts of disease mechanisms and available biomarkers are summarized to help understand achievable outcomes and limitations of cPMP substitution.
Synthesis of cyclic pyranopterin monophosphate and molybdoenzymes in humans
Humans require molybdenum (Mo) as a catalyst for four oxidoreductases. The cell biology of molybdenum and the synthesis of the molybdenum cofactor (MoCo) have been reviewed in detail elsewhere.1,2,3 In short, the synthesis of mature MoCo requires four steps, involving substrate transport across membranes, and enzyme complexes in mitochondria and the cytosol (Figure 1), which are encoded by four genes with multiple gene products (Figure 2).
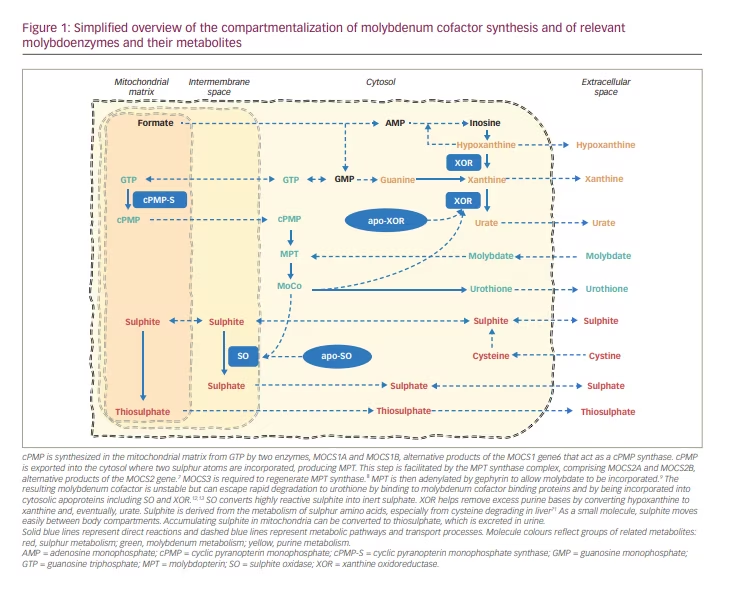
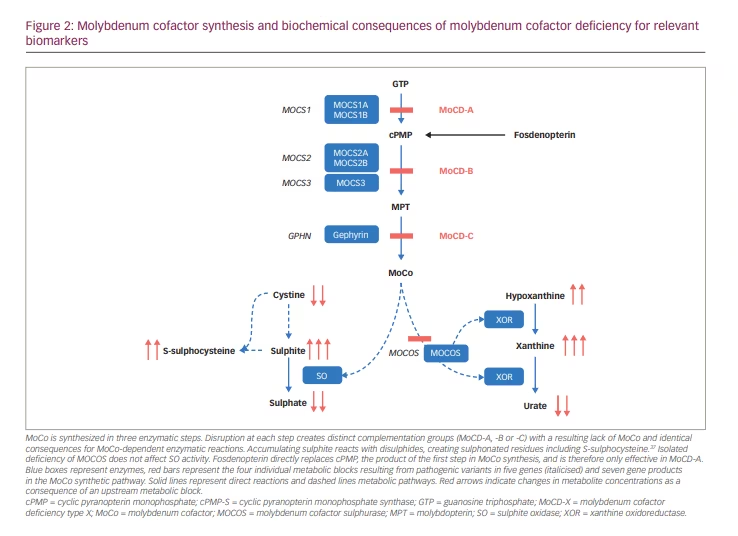
cPMP was identified in 1993 as Precursor Z, the first and most stable intermediate in MoCo synthesis.4 When it was confirmed that Precursor Z exists in the cyclo-pyrano form, the alternative name cPMP was proposed.5 cPMP is a tricyclic pteridine molecule that is synthesised in the mitochondrial matrix directly from guanosine-5’-triphosphate (GTP) by two enzymes, molybdenum cofactor synthesis (MOCS)1A and MOCS1B, which are alternative products of the MOCS1 gene.6 cPMP is exported into the cytosol where two sulphur atoms are incorporated, producing molybdopterin (MPT). This step is facilitated by the MPT synthase complex, comprising MOCS2A and MOCS2B, alternative products of the MOCS2 gene.7 MOCS3, encoded by the MOCS3 gene, is required to regenerate MPT synthase.8 Gephyrin, encoded by the GPHN gene, facilitates the adenylation of MPT and subsequent insertion of molybdate with cleavage of adenylate to form the mature MoCo.9
cPMP, MPT and MoCo are all sensitive to oxidative inactivation. MPT and MoCo have a very short biological half-life in oxidative environments.10 MoCo is protected against oxidation to Form B and enzymatic degradation to urothione by binding to specific binding proteins11 or by incorporation into one of the apoproteins of the four known human molybdoenzymes.12,13 The mature MoCo is directly incorporated as a prosthetic group into the nascent apoproteins of sulphite oxidase and of the mitochondrial amidoxime reducing component. The incorporation of MoCo is effectively required for mitochondrial import and retention of sulphite oxidase (SO).12 In contrast, MoCo must be modified by MoCo sulphurase before it can bind as cofactor to the apoproteins of xanthine oxidoreductase (XOR) or aldehyde oxidase (AO).14
Clinical presentation of molybdenum cofactor deficiency
Defects in MoCo synthesis lead to deficiencies of all molybdoenzymes, except for defects in MoCo sulphurase, which only affect XOR and AO (Figure 2). By far, the most relevant consequence of MoCD is the resulting lack of SO activity. Isolated sulphite oxidase deficiency (ISOD), due to genetic variants in the SUOX gene, causes a distinctive infantile neurological syndrome that was first described in 1967.15,16 MoCD was first described in 1978.17 Clinically, ISOD and MoCD present with the same symptoms and complications caused by sulphite accumulation, except those patients with MoCD can, in addition, develop nephrolithiasis due to impaired XOR activity.18
MoCD and ISOD are ultra-rare diseases, with published reports of fewer than 200 patients with MoCD19–21 and fewer than 100 with ISOD.22 Biallelic pathogenic variants in MOCS1 impair the production of cPMP and are classified as MoCD type A (MoCD-A), which is the most commonly diagnosed subtype of MoCD. Most other cases are MoCD type B (MoCD-B), caused by MOCS2 variants23 or, rarely, MOCS3 mutations.24,25 Only a few cases of MoCD type C (MoCD-C) (caused by GPHN mutations) have been found.26 There are no published data on the incidence of MoCD available. From the author’s own unpublished observation, the prevalence at birth is at least 1 in 200,000 newborns in the UK.
Pre-natal brain abnormalities have been reported in a small number of infants with MoCD, including subcortical cysts, dysgenesis of the corpus callosum,27 as well as enlarged lateral ventricles and, more commonly, an enlarged cisterna magna and cerebellar hypoplasia.28,29 These abnormalities indicate that early brain injury occurs in some cases from mid-gestation onwards.29 Mild brain oedema has been identified in some foetuses from 36 weeks of gestation,28 leading to consideration of early delivery for affected infants.28,30 Protection from accumulating sulphite through trans-placental maternal clearance30,31 ceases at birth, resulting in fulminant encephalopathy in infants with severe MoCD, which manifests within hours to days after delivery. Clinical signs are feeding difficulties, irritability, exaggerated startle reactions, decreased consciousness, apnoea, seizures, axial hypotonia and limb hypertonia.18 Newborns can manifest with moderate metabolic acidosis and increased lactate, as well as with hypoglycaemia. Other routinely performed biochemical investigations are typically normal. Brain magnetic resonance imaging (MRI) at this early stage of disease reveals generalized oedema and widespread restricted diffusion, indicating neuronal damage.20 The presentation is similar to that of hypoxic brain injury, for which it may be mistaken.
After the neonatal period, surviving infants develop a relatively uniform phenotype. They continue to be irritable, have myoclonic and generalized seizures, and develop severe spastic cerebral palsy with intermittent dystonic crises. They typically have profound developmental delay and their brain is atrophic with acquired porencephaly and microcephaly. Children are at high risk of secondary complications from seizures, aspiration, and lower respiratory tract infections. Older children display marfanoid features, including lens dislocation, and a few cases of urolithiasis due to xanthinuria have been reported.18,32 Their reported median age at death is between 2.4 years21 and 3.0 years,19 and is dependent on the severity of illness but also on the extent of medical care provided.
Attenuated disease manifestations are being increasingly reported and likely represent a milder biochemical defect. Symptom onset may occur during infancy or childhood and can be of insidious or sudden onset, with dystonia, spasticity, and varying degrees of developmental delay.33,34 There is a genotype-phenotype correlation; a few MOCS1 and MOCS2 variants are associated with later disease onset and milder clinical manifestations,21,35 including less pronounced biochemical abnormalities. There is, however, no overlap between healthy individuals and those with untreated MoCD.21
Biochemical derangement in molybdenum cofactor deficiency and clinically relevant biomarkers
Most known cases of MoCD are of neonatal onset and are associated with severe biochemical disturbance and disease, including early acute encephalopathy and seizures. MoCD-A and MoCD-B largely overlap in their clinical and biochemical features and can only be distinguished by genetic assessment or measuring urinary cPMP concentrations in a research laboratory.26, 36
SO converts highly reactive sulphite into inert sulphate (Figure 2).15 SO deficiency leads to sulphite accumulating, resulting in a multitude of secondary biochemical abnormalities. As a small molecule, sulphite readily moves between body compartments and is found in increased concentrations in all body fluids.18 Increased sulphite in urine can be easily detected using sulphite test strips; however, these are not licensed for medical use and can yield false positive and false negative results.18,26 In mitochondria, sulphite can be converted to thiosulphate, which is excreted in urine. Sulphite readily reacts with disulphides, including with free L-cystine, yielding S-sulphocysteine (SSC), which is also renally excreted.37 Thiosulphate and SSC are reliable and stable markers of sulphite accumulation that rise very quickly after birth and can be measured in blood or urine in specialized laboratories. SSC concentrations show great interindividual but little intraindividual variability over time.21
XOR is involved in removing excess purine bases by converting hypoxanthine to xanthine and eventually to urate (Figure 2). The lack of XOR activity causes xanthine and hypoxanthine to accumulate, as well as greatly reducing urate production. Plasma urate is in equilibrium with the maternal circulation during pregnancy, and plasma concentrations in neonates with MoCD can remain within the normal range during the first few days of life until renal clearance has removed all maternal urate from their circulation.18 Plasma urate is normal in ISOD and can be decreased but present in milder MoCD.21 Plasma or urinary purine metabolites are a more sensitive marker of compromised XOR activity and can be measured in specialized metabolic laboratories.18
Current hypotheses of pathomechanisms in molybdenum cofactor deficiency
MoCD is a primarily neurological disease affecting neurons in the central and peripheral nervous system. The disease course suggests acute neuronal energetic failure and research has shown that mitochondrial energy metabolism is directly impaired by accumulating sulphite.38 Sulphite increases oxidative stress and reduces adenosine triphosphate (ATP) synthesis by directly inhibiting glutamate hydrogenase in mitochondria that respire on glutamate hydrogenase.39 Sulphite can disrupt mitochondrial integrity and function,40 and mitochondrial respiration was found to be impaired in a MOCS1 defective cell line41 and in cells of SO-deficient mice.42 In addition, a specific pathomechanism has been proposed that can explain the apparent peracute neuronal failure after birth: SSC is a strong N-methyl-D-aspartate (NMDA) receptor agonist,43,44 and leads to excitotoxic neuronal cell death and seizures.45–47 It is therefore likely that, in MoCD, the encephalopathic crisis after birth is caused by an excitotoxic storm that leads to widespread neuronal necrosis.
Cleavage of protein disulphide bridges by sulphite alters the tertiary structure and function of enzymes and structural proteins.37 Cleavage of such structural proteins disrupts connective tissue, causing marfanoid features and lens dislocation.18
Direct inhibition of the enzyme alpha-amino adipic semialdehyde (AASA) by sulphite has been demonstrated and leads to a secondary deficiency of bioavailable pyridoxal 5′-phosphate, which may contribute to hyperexcitability and seizure activity.48, 49
Development of cyclic pyranopterin monophosphate treatment for molybdenum cofactor deficiency type A
Preclinical work
Until recently, there was no causal treatment for MoCD. Affected children have benefitted from symptomatic and supportive treatment. Sulphite accumulation can be reduced with dietary restriction of sulphur-containing amino acids,50 but any attempts to modify the course of disease in severe MoCD, for example with administration of ammonium molybdate, sulphate, thiamine, D-penicillamine, mesna and tetrahydrobiopterin, were unsuccessful. Supplementation with pyridoxine has been suggested in milder cases of MoCD.51
Replacing the lacking MoCo has been tried for decades. Directly replacing isolated MoCo or MPT is hampered by their lability in aerobic environments. However, co-cultivation of cells from different MoCD patients could restore the concentration of MPT and activity of molybdoenzymes in some cell lines, suggesting the presence of a diffusible intermediate in cofactor synthesis and the existence of a stable precursor to MPT.52 Although this precursor was identified in 1993,4 it took another 10 years to establish its exact structure.5 Creating a transgenic MOCS1 defective mouse model,53 and the recombinant overexpression of cPMP-producing enzymes in Escherichia coli eventually allowed testing the previously proposed substrate replacement with cPMP in vivo. E. coli-derived and purified cPMP (rcPMP) given to MOCS1 -/- mice within 5 days after birth and then every 3 days per transabdominal intrahepatic injection ensured survival and normalisation of xanthine and SSC. Liver MPT levels reconstituted to maximally 16% of wild type on the day of injection. SO activity reached a maximum of 26% and XOR reached around 40% of wild-type activity on day 1 after injection. Stopping the treatment led to behavioural deterioration after 6 days of withdrawal.54
First-in-human use and administration of cyclic pyranopterin monophosphate on named patient basis
The administration of rcPMP to humans with MoCD started in 2008 on a named patient basis as an experimental treatment and according to a strict prospective observational protocol designed by Orphatec Pharmaceuticals GmbH (later renamed Colbourne Pharmaceuticals GmbH), a spin-off from the original research group.55 The first child with MoCD-A was 36 days old when treatment started, and responded within a few days with normalized biomarkers and clinical improvement. However, the sequelae of post-natal neuronal injury could not be averted, and the child later showed typical signs of severe cerebral palsy.36 The second infant with MoCD-A was treated in 2009 under similar circumstances but treatment was started earlier, at the age of 7 days, and prior to developing seizures. This child showed normalized biomarkers and normal early development.56 A third child was treated in 2010 from the age of 5 days. This child was already severely encephalopathic at the start of treatment and, while biomarkers improved as expected, the child developed typical sequelae of brain injury and severe cerebral palsy.56,57 A further two neonates were treated in 201058 and 2011, within hours after birth, and both showed moderate developmental delay in childhood.28 No adverse reactions to treatment were observed under continued cPMP administration for these and a few other children.59
In September 2010, prompted by these encouraging results, the European Commission granted orphan designation for cPMP for the treatment of MoCD-A to Orphatec Pharmaceuticals GmbH.60 In 2011, Alexion Pharma International Sarl acquired the cPMP programme and started chemically synthesizing cPMP. The synthetically derived cPMP, fosdenopterin, has identical properties to the naturally occurring rcPMP.61 In March 2013, the European Medicines Agency transferred sponsorship to Alexion Europe SAS, who also received breakthrough therapy designation from the US Food and Drug Administration (FDA) in the same year. Origin Biosciences Inc acquired the late stage cPMP therapy programme in 2018 and, in February 2021, the FDA approved fosdenopterin for the treatment of MoCD-A in the USA.62
Clinical trials and treatment experience with cyclic pyranopterin monophosphate
The circumstances and outcomes of the first cohort of 16 infants treated with recombinant cPMP on a named patient basis and following a prospective monitoring protocol were published in 2015.59 Five of those infants were siblings of previous index cases, including one child with MoCD-B. Eleven others were diagnosed based on their symptoms after birth and, of those, seven were affected with MoCD-A and four with MoCD-B. Treatment in all five infants with MoCD-B was terminated after lack of response to cPMP substitution. Treatment was also discontinued in five of the 11 infants with MoCD-A who showed a biochemical response but whose disease had progressed to an extent that cPMP substitution was deemed futile by the treating physicians and parents.
From 2011, Alexion Pharma International Sarl had designed and sponsored a series of international clinical trial protocols to develop cPMP therapy (Table 1),63-66 which were later continued by Origin Biosciences Inc. Studies ALX-MCD-501 (NCT01640717)63 and ALXN1101-MCD-101 (NCT01894165)64 have been completed. The trials ORGN001-MCD-201 (NCT02047461, EudraCT 2013-002701-56)65 and ORGN001-MCD-202 (NCT02629393, EudraCT 2013-002702-30)66 are ongoing. Data from these trials have not yet been made publicly available, apart from excerpts and summaries contained in the prescribing information for fosdenopterin hydrobromide dihydrate (Nulibry™, Origin Biosciences, Boston, MA, USA)67 and a preliminary poster presentation at a scientific meeting.68
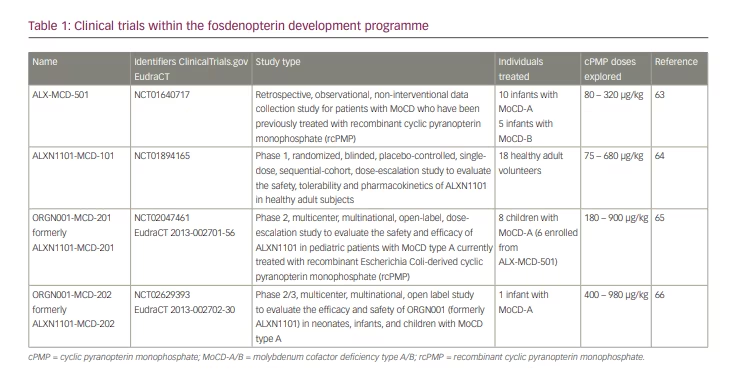
Six patients from the original cohort and two additional patients who were treated later remained on rcPMP treatment until they were switched to fosdenopterin. This switch was monitored under clinical trial protocol ORGN001-MCD-201, which also included a dose escalation exercise with pharmacokinetic and pharmacodynamic assessments.65 Another patient was started on fosdenopterin under the trial protocol ORGN001-MCD-202,66 bringing the total of long-term treated patients to nine. Survival of all infants treated with cPMP was improved compared to matched untreated controls.67
Dosage of cyclic pyranopterin monophosphate
Intravenously administered cPMP has an elimination half-life of 1.2–1.7 hours, with a proportion of renal clearance of around 40%.67 A large proportion of cPMP is oxidized non-enzymatically to compound Z, and a smaller proportion is converted to MPT and eventually to MoCo. Some mature MoCo is incorporated into molybdoenzymes and some degrades to Form B or urothione, which are lost in urine. Estimated daily molybdenum requirements for healthy adults are 25 µg.69 The European Society for Paediatric Gastroenterology Hepatology and Nutrition and The European Society for Clinical Nutrition and Metabolism recommend 0.25 µg/kg/day of molybdenum in term infants and children receiving long-term parenteral nutrition.70 Assuming this is sufficient to produce adequate amounts of mature MoCo, equivalent cPMP requirements would be around 100 µg/day in adults and 1–2 µg/kg/day in infants. Pharmacological dosing requirements of the precursor cPMP are dependent on mode and frequency of administration and are likely higher when cPMP is given as intermittent intravenous boluses. Schwarz et al. suggested an intravenous treatment dose for cPMP of 100 µg/kg twice weekly, based on their original mouse experimentation and the observed biological half-life of holo-molybdoenzymes,54 which is a highly relevant parameter to consider when establishing dosing intervals.
The first few patients were started on a dose of 80 µg/kg cPMP once daily, administered in staggered short intravenous infusions, and the dose was sequentially escalated over 2–3 months to 240 µg/kg once daily.56 Later patients were started on a dose of 240 µg/kg/day. 59 The licensed starting dose for fosdenopterin is 400 µg/kg in pre-term infants and 550 µg/kg in term infants, both escalating to 900 µg/kg once daily from month 3 of treatment and from the start of treatment for any children over the age of 1 year.67 cPMP substitution in the doses used so far can restore normal concentrations of biomarkers.59 Based on studies of the mouse model of MoCD-A, it is likely that the current dosage is not sufficient to restore full enzymatic activity.54 This may, however, not be required to prevent harmful sulphite or xanthine accumulation.
Safety of cyclic pyranopterin monophosphate
The safety of cPMP has been demonstrated since 2008 with daily administration of rcPMP over approximately 3 years in eight patients, and of fosdenopterin hydrobromide dihydrate over a median duration of 4.3 years in nine patients. No immune-mediated infusion-associated reactions or direct toxic effects have been observed. Adverse events reported during clinical trials related to intercurrent illnesses of childhood and complications associated with central venous access. Daily intravenous administration normally requires a surgically placed, tunnelled intravenous line, which incurs a risk for typical complications, such as exit site infection, septicaemia, or catheter blockage. One infant died from necrotising enterocolitis while treated with cPMP.67
Animal studies have identified a potential risk of ultraviolet phototoxicity in patients receiving high doses of fosdenopterin.67 Patients on cPMP substitution therapy should avoid or minimize exposure to sunlight or artificial ultraviolet light. The number of patients exposed to fosdenopterin is still very small and further safety surveillance is required to determine the relevance of phototoxicity in clinical practice.
Biochemical efficacy of cyclic pyranopterin monophosphate
Available data demonstrate good and sustained biochemical efficacy with any of the previously used dosing regimens.59 In MoCD-A, a decrease in sulphite, SSC, xanthine and hypoxanthine is expected within 24 hours and an increase in urate in body fluids within 24–48 hours of initiating treatment. Previously grossly abnormal biomarkers remain close to or within the reference range under treatment.59 There has been no tachyphylaxis reported in any of the long-term treated patients for over 10 years. Pharmacodynamic data from the clinical trials have not been published, and the optimum dose and administration intervals have not been robustly established yet.
Long-term clinical outcomes of children treated with cyclic pyranopterin monophosphate
The clinical outcomes of treated patients have been variable. At first look, this may be surprising, given the uniformly good biochemical response to cPMP substitution in patients with MoCD-A. The clinical outcome, however, does not solely depend on the biochemical response of the patient. The neurological outcome of treated infants correlates closely with the state of consciousness at initiation of treatment, limiting relevant developmental progress to those who were treated prior to having entered a comatose state.59 Correcting the biochemical abnormality in MoCD-A is expected to protect injured and intact neurons, but cannot reverse any preceding neuronal necrosis. This implies that the neurological outcome of patients mainly depends on the extent of initial brain injury sustained prior to intervention.
A few children with severe MoCD-A have been treated prior to experiencing seizures or becoming comatose and some have been treated immediately after birth. While these patients have not developed cystic encephalomalacia, brain atrophy with secondary microcephaly, or cerebral palsy, and some have never experienced any seizures, sequelae such as speech delay, mild learning difficulties and mild central muscular hypotonia have been observed.28,59 This indicates an incomplete treatment effect, either due to subclinical pre- or immediately post-natal brain injury or due to an ongoing disease process that is not reflected in the concentration of currently used plasma and urine biomarkers.
Children whose treatment started after having suffered significant brain injury have continued to have seizures, frequent myoclonus and severely impaired motor and cognitive function, as well as the typical neuroradiological sequelae of MoCD. However, with ongoing cPMP treatment, their survival is improved over a comparable untreated group21,67 and their risk of nephrolithiasis due to xanthinuria is abolished due to a normalisation of urinary xanthine concentrations. It is difficult to quantify other presumed benefits of treatment, such as reduced irritability and an improvement in their families’ quality of life.
Children with milder MoCD-A and late symptom onset are likely to benefit from cPMP substitution, which may prevent episodes of acute neurological deterioration and improve cognitive developmental progress.
Practical considerations and unmet needs
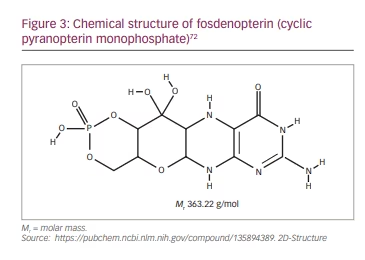
Fosdenopterin is supplied as a sterile, preservative-free, lyophilized powder in a single-dose, clear glass vial for reconstitution. Each vial contains 9.5 mg fosdenopterin (equivalent to 12.5 mg fosdenopterin hydrobromide as a dihydrate).67 It should be noted that the molar mass of cPMP is 363.22 g/mol (Figure 3) and that of fosdenopterin monobromide dihydrate is 480.16 g/mol. After reconstitution, the final concentration is 9.5 mg fosdenopterin in 5.0 mL (1.9 mg/mL) and the dose is administered undiluted through a 0.2 µm filter as a short intravenous infusion, at a rate of 1.5 mL/min. Fosdenopterin must be stored frozen between -10°C and -25°C and used within 4 hours after reconstitution.
From the author’s personal observation, the requirement for daily intravenous administration puts a significant burden on carers of affected children who administer treatment at home after the initial hospitalization. This requires resources in terms of healthcare provision, space at home and time of carers, and it limits the freedom of movement for the families. While all this can and has been overcome successfully in several cases, more research into dosing intervals and alternative modes of application of fosdenopterin is required to make this treatment more widely available and reduce the risk-benefit ratio.
Timely diagnosis of affected individuals is the greatest challenge. Severely affected neonates may become symptomatic within hours to days after birth, and irreversible brain injury occurs rapidly.58,59 The initial presentation, especially in late-onset cases, is unspecific, and reliable testing for sulphite accumulation is not always readily available.33 However, the option of treating children on suspicion of MoCD and prior to diagnostic confirmation is within the licensed application of fosdenopterin. Ideally, a validated bedside test that can detect elevated sulphite and xanthine should be developed to allow timely diagnosis and treatment. Further delays may be incurred if fosdenopterin must be transported to the patient.
While fosdenopterin is an excellent treatment for MoCD-A, other strategies are required to provide treatments for the closely related MoCD-B and MoCD-C, as well as for ISOD.
The following recommendations are based on the author’s personal experience. Treatment with cPMP should include close clinical surveillance and initial frequent monitoring of biomarkers, such as xanthine, urate, sulphite and SSC to establish a biochemical response, especially if cPMP substitution is started prior to genetic diagnosis. Measurements of SSC and purines in urine or plasma may not always be available in a timely fashion. Commercially available sulphite dip sticks are not licensed for medical use and, therefore, are not generally accepted for monitoring purposes. Plasma urate is a more commonly used assay, and results are expected to be available in most neonatal and paediatric units within 24 hours. An increase in plasma urate within 2 days of cPMP substitution indicates a treatment response.
Conclusion
cPMP substitution is a safe and effective causal treatment for MoCD-A that can prevent disastrous disability and early death. The benefits for the child’s development are remarkable when treatment is started in pre-symptomatic children and justify the burden of treatment. The disease-modifying benefit of fosdenopterin treatment is less clear in children whose treatment is started after they have suffered irreversible neuronal injury.
A high index of suspicion and easy access to diagnostic tests and the drug are crucial to ensure good clinical outcomes. Given its good safety profile, early empirical fosdenopterin treatment of encephalopathic infants with a possible diagnosis of MoCD should be considered.
The requirement for frozen storage and daily intravenous administration of fosdenopterin is challenging in day-to-day practice. Surveillance of treatment outcomes and further research into alternative modes of drug delivery will aid the wider implementation of this novel treatment.