Neuromyelitis optica spectrum disorder (NMOSD) is an inflammatory disease of the central nervous system (CNS) and is known to cause recurrent episodes of optic neuritis and transverse myelitis. Myelitis is radiologically referred to as longitudinal extensive transverse myelitis on spinal cord magnetic resonance imaging (MRI), extending to ≥3 vertebral segments. The disease flare, either at onset or during relapse, is often more serious in NMOSD than in multiple sclerosis (MS), a representative demyelinating disease. In fact, the occurrence of even a single relapse may cause persistent disabling symptoms, implying that prevention of relapse has more significant meaning in NMOSD than in MS. After the discovery of an antibody that binds to the aquaporin-4 (AQP4) water channel,1 experts soon realized the remarkable differences between patients with MS and those with anti-AQP4 antibody (AQP4-Ab) positivity, who are now operationally classified as patients with ‘seropositive NMOSD’. Although the diagnosis of neuromyelitis optica was previously given to limited patients regardless of AQP4-Ab seropositivity, the committee of the 2015 criteria recommended the diagnostic term of NMOSD, which covers broader seropositive and AQP4-Ab-seronegative patients.2
To diagnose seropositive patients, the confirmation of at least one of the six core characteristics, including optic neuritis, acute myelitis, area postrema syndrome (unexplained hiccups, nausea or vomiting), acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome, and symptomatic periependymal cerebral syndrome, is needed. The exclusion of alternative diagnoses is also required. For the diagnosis of seronegative patients, stricter criteria are applied, which include two of the six core clinical characteristics (as listed above), MRI-confirmed lesions, and exclusion of alternative diagnoses.2 However, further research and discussion are needed for the identification of patients with seronegative NMOSD. Recently, antibodies against myelin-oligodendrocyte glycoprotein (MOG) have been reported to be associated with a proportion of seronegative NMOSD.3 MOG is a myelin antigen, and anti-MOG-positive patients have a broader spectrum of diseases.
Serious acute relapses in patients with NMOSD and MS are treated with intravenous high-dose corticosteroids and plasma exchanges. However, disease-modifying therapies approved for MS are not recommended for the treatment of NMOSD as the use of some disease-modifying drugs may be harmful to patients with NMOSD.4 Thus, oral corticosteroids and general immunosuppressants (such as azathioprine and mycophenolate mofetil), or an anti-CD20 monoclonal antibody (e.g. rituximab), have been prescribed in the remission phase of NMOSD. However, recent studies have identified various therapeutic targets in the molecular pathways of NMOSD, leading to the development of therapeutic monoclonal antibodies such as satralizumab, the efficacy of which has recently been demonstrated in randomized clinical trials.5–8 This narrative review explores the involvement of IL-6 in NMOSD and the potentials of satralizumab, a humanized antibody against IL-6R, in the current and future practice of NMOSD.
Neuromyelitis optica spectrum disorder as an autoimmune disease involving interleukin-6
The discovery of AQP4-Ab has led to a better understanding of the pathophysiology of NMOSD. Histopathological analysis has demonstrated that loss of AQP4 expression in astrocytes is associated with complement deposition in the CNS of patients with NMOSD, which occurs prior to the destruction of astrocytes.9 AQP4 loss appears to be independent of primary demyelination, which supports the hypothesis that AQP4-Ab and the complement system play a pivotal role in the pathogenesis of NMOSD.
As a reflection of the inflammatory pathology of NMOSD, previous studies have documented the elevation of cytokines and chemokines in the serum and cerebrospinal fluid (CSF) of patients.10 Notably, the levels of interleukin (IL)-6, a pro-inflammatory cytokine, in the serum of patients with NMOSD were higher than in the serum of patients with other neurological diseases, including MS,11 and were found to predict the occurrence of early and severe relapses.12 IL-6 is a pleiotropic cytokine that induces the synthesis of acute inflammatory phase proteins, such as C-reactive protein, serum amyloid A and fibrinogen from hepatocytes, and inhibits the production of albumin.13 It stimulates antibody production and induces the differentiation of effector T cells. Moreover, it promotes the activation and development of some non-immune cells, such as fibroblasts and platelets. Thus, persistent production of IL-6 may trigger the onset or exacerbation of immune-mediated diseases.
A pioneering study by Hirano et al. initially identified IL-6 as a cytokine that induces B-cell production of immunoglobulin.14 A subsequent study identified various roles of IL-6 and demonstrated how this cytokine transmits signals that lead to various biological outcomes. The binding of IL-6 to cell surface IL-6 receptor (IL-6R) forms the IL-6/IL-6R complex, which induces homodimerization of gp130. IL-6 binding to soluble IL-6R also induces gp130 homodimerization. This event critically triggers activation of the Janus kinase (JAK)-signal transducer and activator of transcription 3 pathway and the JAK-SHP-2 mitogen-activated protein kinase pathway, leading to biological responses.13
In the pathogenesis of NMOSD, IL-6 is thought to play multiple roles, including induction of pathogenic effector T-cell development, promotion of plasmablast survival, production of AQP4-Ab and disruption of blood–brain barrier integrity and functions (Figure 1).15 The CD4+ helper T cell (Th cell) subset plays a key role in the induction of adaptive immune responses and is assumed to serve as the commander in autoimmune diseases. Further, AQP4 peptides from the membrane surface of AQP4 can activate Th17 cells, producing IL-17 in NMOSD.16 This process is influenced by IL-6 as it is critical for the development of Th17 cells and suppresses regulatory T-cell differentiation.17 In a rodent model, IL-6, produced by B cells, played a predominant role in inducing Th17 cells associated with brain inflammation.18 IL-6 and IL-21 are specifically increased in NMOSD compared with MS, whereas other inflammatory proteins, such as IL-17 and IL-1-beta, are increased in both NMOSD and MS.10
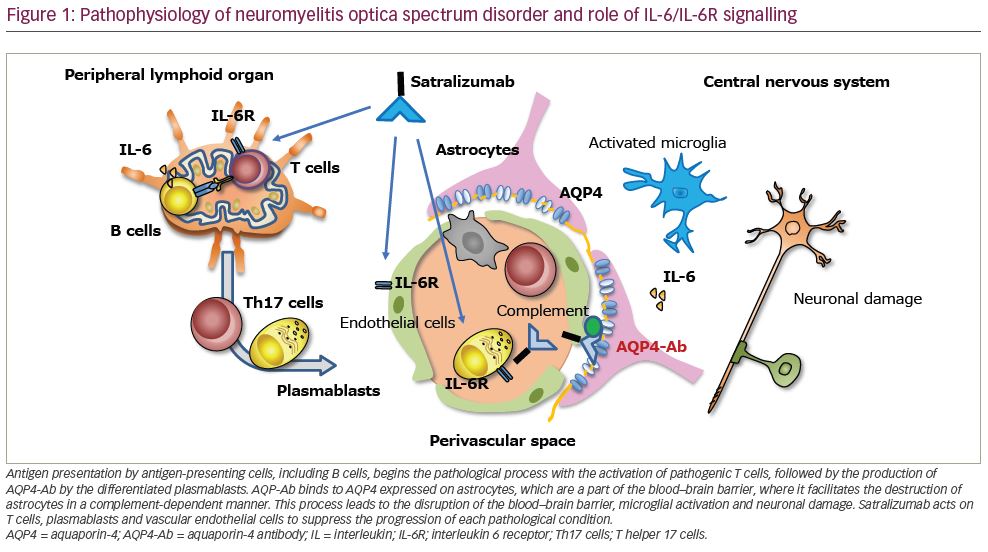
B cells play a role in the pathophysiology of NMOSD, given that B cell-targeted therapies such as those targeting CD20 and CD19 have proven to be efficacious in NMOSD.7,8 As B cells produce IL-6, B cell-targeted therapy may show efficacy by reducing the source of IL-6 involved in NMOSD. However, as B cells serve as antigen-presenting cells for pathogenic Th17 cells, B cell-targeted therapies may dampen Th17 cell activation.
It has been reported that differentiated plasmablasts (CD19lowCD27highCD38highCD180– B cells) are increased in the peripheral blood of patients with NMOSD compared with those in healthy subjects or patients with MS.19,20 Although plasmablasts have eccentric nuclei, perinuclear hof regions and abundant cytoplasm, they have a larger nucleus with a lower extent of chromatin clumping than plasma cells. Thus, it seems that plasmablasts are involved in the process of cell differentiation just before mature plasma cells are located in the niches in the bone marrow.
IL-6 has been reported to promote the survival of plasmablasts derived from patients with active NMOSD.19 The plasmablasts produced AQP4-Ab ex vivo and IL-6 enhanced autoantibody production in plasmablasts. These phenomena were not observed when plasmablasts were stimulated by other B cell-stimulating factors, such as a B cell-activating factor and a proliferation-inducing ligand.19 Moreover, blocking IL-6R signalling by an antagonistic antibody for IL-6R decreased the number of cultured plasmablasts. These observations led to the idea of using an IL-6R-blocking antibody in the treatment of NMOSD.
Importantly, the increase in plasmablasts was shown to be more prominent in the relapse phase, especially in the CSF. These plasmablasts in the CSF express the plasma cell surface protein CD138 and human leukocyte antigen class II. Reflecting their ability to migrate to the CFS, they expressed inflammatory tissue-oriented C-X-C motif chemokine receptor (CXCR) 3, but not bone marrow-oriented CXCR4.21 However, the number of somatic hypermutations was not different between plasmablasts from the peripheral blood and those from the CFS and some clones were commonly observed in both sites.21 Thus, peripherally differentiated plasmablasts probably migrate into the CNS and may be part of inflammation onsite during the flare of NMOSD.
Interleukin-6 receptor blockade for neuromyelitis optica spectrum disorder
Based on the findings associating IL-6 and NMOSD, compassionate use of anti-IL-6R antibody has been considered to mitigate devastating NMOSD resistant to the currently available therapies. Case reports from Japan and Germany have indicated that tocilizumab, a humanized monoclonal antibody against IL-6R, could be useful for treatment-resistant patients.22,23 Anti-IL-6R therapy has shown efficacy in patients with intractable NMOSD resistant to intensive treatment with steroids, immunosuppressants, such as anti-CD20 and plasmapheresis.22,23 In a Japanese patient who was previously treated with interferon-β, severe neuropathic pain, fatigue and disability scores gradually improved.22 Subsequently, two small studies exploring the safety and efficacy of tocilizumab confirmed the efficacy of this treatment, indicating that IL-6R signalling plays a key role in the pathogenesis of NMOSD.23,24 Recently, a clinical trial used tocilizumab monotherapy for the treatment of NMOSD, including 85% seropositivity for AQP4-Abs. In this study, a higher percentage of patients without concurrent autoimmune diseases remained relapse free for 2 years compared with those treated with azathioprine (91% versus 63%).25
Satralizumab: A subcutaneously injectable recycling antibody against interleukin-6 receptor
Satralizumab is a humanized monoclonal antibody that binds to both membrane-bound and soluble forms of IL-6R to block the homodimerization of gp130, which prevents downstream signalling pathways. Satralizumab is designed to dissociate from IL-6R at low pH in the endosome and maintain the affinity of constant regions for the neonatal Fc receptor. After internalization of the antigen–antibody complex in the target cells, the antibody dissociates from IL-6R and is released into the circulation. This results in binding to another IL-6R recycling mechanism that prolongs the elimination half-life of the drug in the blood.26
The efficacy and safety of subcutaneous satralizumab treatment concomitant with baseline immunosuppressants were tested in the SAkuraSky study; a phase III randomized, double-blind, placebo-controlled, parallel-assignment trial with an open-label extension period (Table 1).5 Patients with NMOSD (both seropositive and seronegative for AQP4-Ab) were recruited from 11 countries (34 institutes). Concomitant use of azathioprine (<3 mg/kg/day), mycophenolate mofetil (<3,000 mg/day) or prednisolone (<15 mg/day) as baseline immunosuppressants was allowed. The doses of the baseline treatment drugs were fixed during the study. Patients who recently received B-cell depletion therapy were not included. Of note, not only adult patients (18–74 years) but also adolescents aged 12–17 years were included in the study. Randomization was stratified according to the baseline annual relapse rate (1 versus >1) and geographic region (Asia versus Europe or other). Patients received subcutaneous satralizumab (120 mg/time) or placebo at weeks 0, 2 and 4, and every 4 weeks thereafter during the double-blind period. The primary outcome was the time to first protocol-defined relapse and the relevant secondary outcomes included changes in the visual analogue scale (VAS) score, which measures pain, and the functional assessment of chronic illness therapy-fatigue (FACIT-F) score, assessing fatigue from baseline to week 24. A sample size of 70 evaluable patients was considered based on the assumption of a 66.5% reduction in the risk of relapse compared to placebo, 80% power at the 5% significance level, and a 2-year dropout rate of 10%. This required 26 protocol-defined relapses.
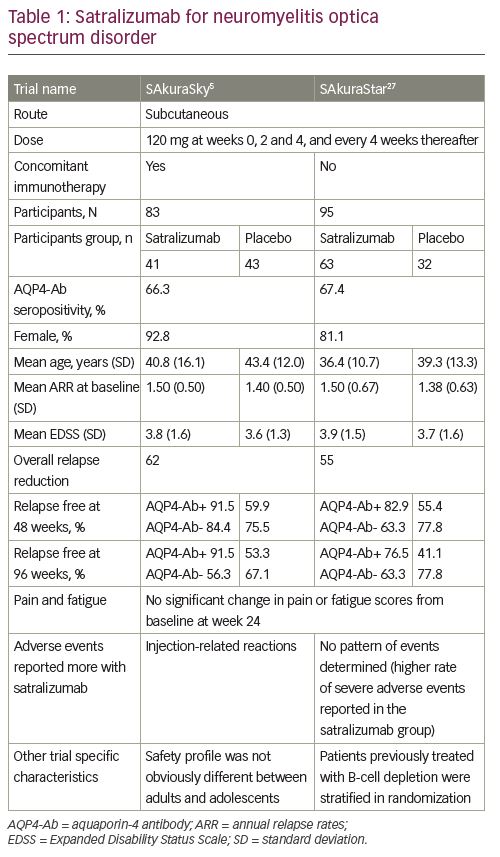
A total of 83 participants were enrolled, and around 40% of patients were Asian residents (39% in the satralizumab group and 43% in the placebo group). In this study, the satralizumab group had a lower risk of NMOSD relapse than the placebo group during the double-blind period. Only eight (19.5%) patients among those receiving satralizumab experienced a protocol-defined relapse, whereas 18 (42.9%) patients among those receiving placebo experienced a protocol-defined relapse (hazard ratio [HR]: 0.38; 95% confidence interval [CI]: 0.16–0.88; adjusted p=0.02; median effectiveness evaluation period: 115.1 weeks for satralizumab and 42.5 weeks for placebo). A higher percentage of patients receiving satralizumab remained relapse free than that of patients receiving placebo (at 48 weeks, 89% versus 66%; at 96 weeks, 78% versus 59%, respectively). Of note, in the AQP4-Ab seropositive subgroup the effect of satralizumab was prominent; a protocol-defined relapse was confirmed in 3 of 27 (11%) patients receiving satralizumab and in 12 of 28 (43%) patients receiving placebo (HR: 0.21; 95% CI: 0.06–0.75). In striking contrast, there was only a small difference in AQP4-Ab seronegative patients between the satralizumab and placebo groups; 5 of 14 (36%) patients receiving satralizumab had a protocol-defined relapse, while 6 of 14 (43%) patients receiving placebo had a protocol-defined relapse (HR: 0.66; 95% CI: 0.20–2.24). There was no significant difference between the groups in terms of change in pain or fatigue scores from baseline to week 24.5
Safety was similar between the adult and adolescent groups. There were no deaths or anaphylactic reactions during the double-blind or evaluation period of satralizumab therapy. Adverse events in the satralizumab group, including injection-related reactions (12%), nasopharyngitis (24.4%), upper respiratory tract infections (24.4%) and headache (24.4%), were also observed in the placebo group. The limitations of this trial included the small group size, the lack of a treatment group of an active comparator and the that fact that long-term safety data are not available. Despite these limitations, this trial showed that satralizumab treatment prolonged the relapse-free period in patients with NMOSD, particularly in patients with seropositivity, but did not demonstrate significant effects on pain or fatigue scores.5
Satralizumab as monotherapy for neuromyelitis optica spectrum disorder
The efficacy and safety of satralizumab treatment as monotherapy were tested in the SAkuraStar study, a phase III, double-blind, placebo-controlled, parallel-assignment trial with an open-label extension period (Table 1).27 Adult patients with NMOSD (both seropositive and seronegative) were recruited from 13 countries (44 institutes). Randomization was stratified by previous therapy for relapse prevention (B-cell depleting therapy versus immunosuppressants or others) and the nature of the most recent attack in the year before screening (initial attack versus relapse for the patient). The procedure and the primary outcome for SAkuraStar were the same as those in the SAkuraSky study and relevant secondary outcomes also included changes in VAS score and FACIT-F score from baseline to week 24. A sample size of 70 evaluable patients was considered based on the assumption of a 75% reduction in the risk of relapse compared to placebo that required 19 protocol-defined relapses.
A total of 95 participants were enrolled and randomized in a 2:1 ratio to satralizumab alone or placebo. Satralizumab treatment had a lower risk of NMOSD relapse than placebo treatment during the double-blind period; a protocol-defined relapse was confirmed in 19 (30.2%) patients among those receiving satralizumab and in 16 (50.0%) patients among those receiving placebo (HR: 0.45; 95% CI: 0.23–0.89; adjusted p=0.02; median effectiveness evaluation period: 95.4 weeks for satralizumab and 60.5 weeks for placebo). A higher percentage of patients receiving satralizumab remained relapse free than that of patients receiving placebo (at 48 weeks, 76% versus 62%; at 96 weeks, 72% versus 51%, respectively). After normalizing all the protocol-defined relapses during the trial period, the annualized relapse rate was calculated as 0.17 in the satralizumab group and 0.41 in the placebo group (95% CI: 0.10–0.26 and 0.24–0.67, respectively). There was no significant difference between the groups in terms of change in pain or fatigue scores from baseline to week 24. Safety was similar between groups. In the AQP-Ab seropositive group, protocol-defined relapse was confirmed in 9 of 41 (22%) patients receiving satralizumab and 13 of 23 (56.5%) patients receiving placebo (HR: 0.26; 95% CI: 0.11–0.63). In the AQP4-Ab seronegative group, 10 of 22 (46%) patients receiving satralizumab and 3 of 9 (33%) patients receiving placebo had a protocol-defined relapse (HR: 1.19; 95% CI: 0.30–4.78).27
There were no deaths or anaphylactic reactions throughout the study period. The most common adverse events encountered with satralizumab were urinary tract infections (17.5%) and upper respiratory tract infections (15.9%), which were also observed in the placebo group. The limitations of this trial included the small sample size and relatively small number of relapses in the placebo control group. Despite these limitations, this study revealed that satralizumab monotherapy in patients with NMOSD delayed the occurrence of relapse and reduced the relapse rate in AQP4-Ab seropositive patients.27
Conclusion
Although general immunosuppressants have been widely used as the first-line treatment for NMOSD, IL-6R-blocking therapy seems to act on multiple phases of the disease process. Two international, randomized, controlled trials have provided clear evidence regarding the effectiveness of satralizumab in preventing disease activity in patients with seropositive NMOSD. As its efficacy in seronegative patients was not evident, the results provoked a discussion about whether seronegative patients could be classified under NMOSD. Importantly, IL-6 is a key immune mediator in which blockade leads to another immune homeostasis. As tocilizumab provides long-term safety and better quality of life for patients with rheumatoid arthritis, satralizumab is a promising immune modulator. Further analysis of these clinical trials and extension studies will provide additional insights into the effectiveness and long-term safety of satralizumab in the treatment of NMOSD.