Dravet syndrome is a rare genetic disorder that affects approximately 1 in 15,700 individuals. It is one of the most severe epilepsy syndromes of early childhood, with high morbidity and mortality rates.1,2 It is characterized by seizures that begin in infancy and can lead to intellectual disability, developmental delays and a range of other neurological problems.3 The SCN1A gene has been identified as the primary genetic cause of Dravet syndrome, with 80% caused by mutations in the SCN1A gene. Research has focused on developing treatments to target this gene.4 In this expert interview, Rajvinder Karda addresses the current status of SCN1A gene-targeting research for the treatment of Dravet syndrome, discusses the potential for future developments in this field and some of the challenges that researchers face in developing effective treatments. She also highlights her current research project to develop a gene therapy for Dravet syndrome, co-funded by Dravet syndrome UK in partnership with Great Ormond Street Hospital Children’s Charity, UK.
Q. Could you give us a brief overview of the SCN1A gene-targeting research project you are running at Great Ormond Street Hospital?
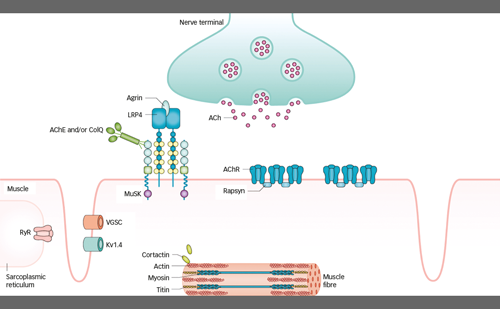
Approximately 80% of people with Dravet syndrome have a loss of function mutation in the SCN1A gene, which encodes a voltage-gated sodium ion channel, Nav1.1.5 This channel is present in brain cells and allows typical firing patterns between these cells. Affected brain cells containing the mutated Nav1.1 channel result in abnormal firing.
Our research project aims to develop a gene therapy treatment for Dravet syndrome. Our approach involves delivering a novel sequence in an adeno-associated virus to increase the healthy SCN1A gene expression and, therefore, restore the Nav1.1 function. The aim is to deliver this treatment to a clinically relevant mouse model for Dravet syndrome, to ameliorate the disease symptoms.
Q. What are some of the key challenges associated with the use of gene-targeting therapies in treating Dravet syndrome?
The key challenges associated with gene-targeting therapies is off-target effects. In our gene therapy treatments, we have overcome this challenge by designing the viral therapy to target the specific cells that express the SCN1A gene.
Q. If successful, how might this gene-targeting therapy impact clinical practice and the lives of people living with Dravet syndrome, and their families?
If our treatment is successful in our preclinical study, this will provide a one-off long-term treatment option for people living with Dravet syndrome and hopefully reduce or completely remove the disease symptoms. Though still at an early stage in the project, the next step following a successful preclinical study would be for clinical studies to be conducted. In addition to the SCN1A gene-targeting research project at Great Ormond Street Hospital, other organizations have treatments for Dravet syndrome that have either started a clinical trial or are due to start one in 2026.6,7
Q. What do you consider the future directions in the development of treatments for Dravet syndrome?
As the field of gene therapy is continuing to develop, this has provided many different forms of long-term effective genetic therapies for Dravet syndrome. This provides the possibility of significantly ameliorating the disease symptoms long term.8–14