Parkinson’s disease (PD) is classically defined as a movement disorder with key features including motor disturbances such as tremor, rigidity, akinesia and postural instability. Non-motor symptoms are also present, and can include disturbances in autonomic function, sleep, cognition, sensory and psychiatric capacity.1 Motor and non-motor symptoms both have a significant impact on quality of life in patients with PD. The pathophysiology of the disease is attributed to progressive degeneration of midbrain dopamine neurons resulting in loss of dopaminergic innervation within the nigrostriatal pathway. Other neurotransmitter systems are likely to be involved in the emergence of non-motor symptomatology. Recent evidence suggests a role for glutamate in the emergence of psychiatric symptoms including depression, anxiety, dementia, sleep and other cognitive disturbances.2,3 Patient progression through early, intermediate and advanced stages of PD demonstrates different pathological underpinnings and imposes a continued challenge, particularly when determining the appropriate therapeutic recommendations.
Dopamine replacement
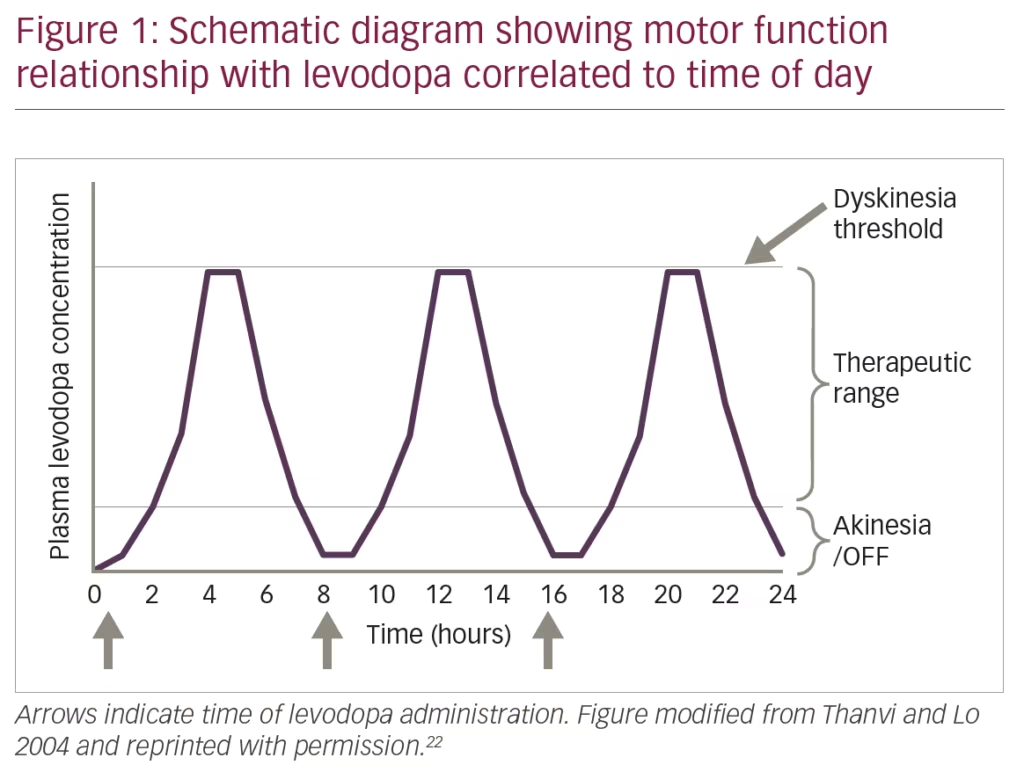
Treatment for PD is largely symptomatic, with dopamine replacement therapy considered the gold standard. Levodopa (L-DOPA), the precursor of dopamine, is the most effective drug in treating the cardinal symptoms of PD but has been associated with the development of significant motor complications following prolonged use.4 These motor complications include dyskinesia and motor fluctuations (i.e., ON/OFF phenomena). L-DOPA-induced dyskinesias comprise abnormal, involuntary motor movements. The ON/OFF phenomena refer to fluctuations in the psychomotor state of patients with PD treated with L-DOPA. These cyclic switches between mobility and immobility can occur towards the end of dosing or as sudden and unpredictable motor fluctuations.5
Community-based studies examining populations of patients with PD that started treatment with L-DOPA before dopamine agonists show that after 5–10 years of treatment, a high frequency of patients developed motor fluctuations (70%) or dyskinesias (60%). In those studies, after 10 years of treatment, an estimated 95% of patients report any type of motor complication,6,7 constituting a major cause of disability in some patients with advanced PD (Figure 1). These complications may be mild at initial presentation but can significantly interfere with quality of life and typically become worse with disease progression, extended time on the drug and dose escalation. The pathogenesis of these complications is complex, and different neurotransmitters such as dopamine, glutamate, adenosine, and gamma-aminobutyric acid (GABA) are suggested to play an important role altering the normal physiology of direct and indirect pathways of the cortico-basal ganglia-thalamic loop responsible for motor control.8–11
The precise mechanisms underlying the development of these complications are largely debatable. Motor complications are attributed in part to the natural progression of the disease, cumulative exposure/dosing of L-DOPA and, more recently, the short half-life of L-DOPA and the unstable, non-continuous dosing regimens which result in pulsatile stimulation of dopamine receptors.12–14 Delayed initiation of L-DOPA therapy gained wide acceptance following reports from several clinical trials in early PD using dopamine agonists that showed significantly reduced risks of developing motor problems when compared with L-DOPA monotherapy.15–17 A recent randomised, delayed-start trial of L-DOPA corroborates these findings and suggests that L-DOPA does not have any disease-modifying effects.18 Different studies have confirmed that the appearance of motor complications following initiation of L-DOPA is also associated with disease duration and L-DOPA daily dose, but not with the time when L-DOPA therapy is initiated.
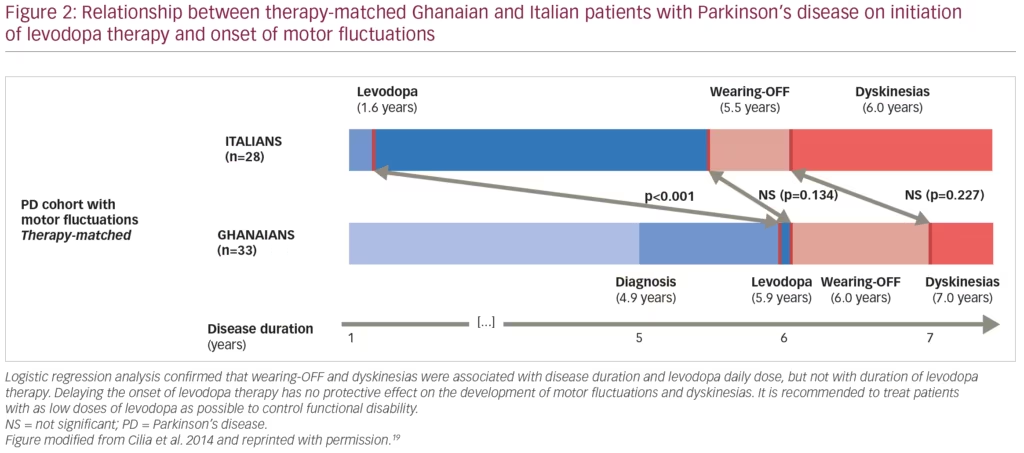
A recent 4-year multicentre study investigated a large cohort of patients with PD in Ghana (where there is limited access to medication and L-DOPA initiation is often delayed after PD onset) and compared outcomes to a control group of patients with PD in Italy where L-DOPA is introduced earlier (mean disease duration 4.2 ± 2.8 versus 2.4 ± 2.1 years).19 The report showed that patients on higher doses of L-DOPA had an accelerated development of motor complications (Figure 2).19 It is also worth noting that the mean disease duration at the baseline visit was 5 years and approximately 35% of patients with PD from Ghana were drug-naïve, a frequency 4-fold higher than in the Italian (control) group. This could contribute to the rapid development of complications, suggesting that a state of continuous dopaminergic denervation may lead to striatal maladaptive compensatory mechanisms. Altogether, these recent studies suggest that delaying the onset of L-DOPA therapy has no protective effect on the development of motor fluctuations and dyskinesias.18,19 We conclude that motor fluctuations and dyskinesias are not associated with the duration of L-DOPA therapy, but rather with longer disease duration and higher L-DOPA daily dose. Hence, the practice to withhold L-DOPA therapy with the objective of delaying the occurrence of motor complications is not justified.
Selective direct-acting dopamine agonists have served as effective monotherapies for the earlier stages of PD, and as adjunct therapies to L-DOPA treatment as the disease progresses. Treatment with dopamine agonists in the short term can reduce the risk for motor complications and also helps to lower the dosage of L-DOPA throughout the course of the disease. Additionally, improvements in non-motor symptoms such as depression have been observed.20 Therefore, combination therapy is beneficial if total L-DOPA dose is reduced, thereby increasing the time that patients are free of disabling motor complications and simultaneous improvement in non-motor symptoms are seen.20,21 One significant caveat in the use of direct-acting dopamine agonists is the development of impulse-control disorders.
As PD symptoms become increasingly refractory to conventional treatments and non-motor symptoms become more significant, it is in the patient’s best interest to explore additional treatment options that may provide enhanced symptom control. Longitudinal management of motor complications is typically approached with dose escalation and fragmentation of L-DOPA as well as adjunct therapies including dopamine agonists, monoamine oxidase-B (MAO-B) and/or catechol-O-methyltransferase (COMT) inhibitors.22,23 Emerging evidence suggests that dysfunction outside of the dopamine system may contribute to motor problems, as well as the development of non-motor symptoms.8–11 Therefore, exploration into therapeutic options that target non-dopaminergic systems have been of interest and provides an adjunct approach to address the limitations of current dopamine-centred approaches.
Glutamatergic modulation
The basal ganglia are a collection of interconnected subcortical nuclei that help to quickly, and precisely, react to external stimuli. The degeneration of dopamine nigral neurons leads to dopaminergic denervation within the striatum causing subsequent functional modifications in the activity of the basal ganglia. The dopamine system is not the only compromised neurotransmitter pathway in this system that is thought to contribute, at least in part, to the underlying pathology of PD (Figure 3).
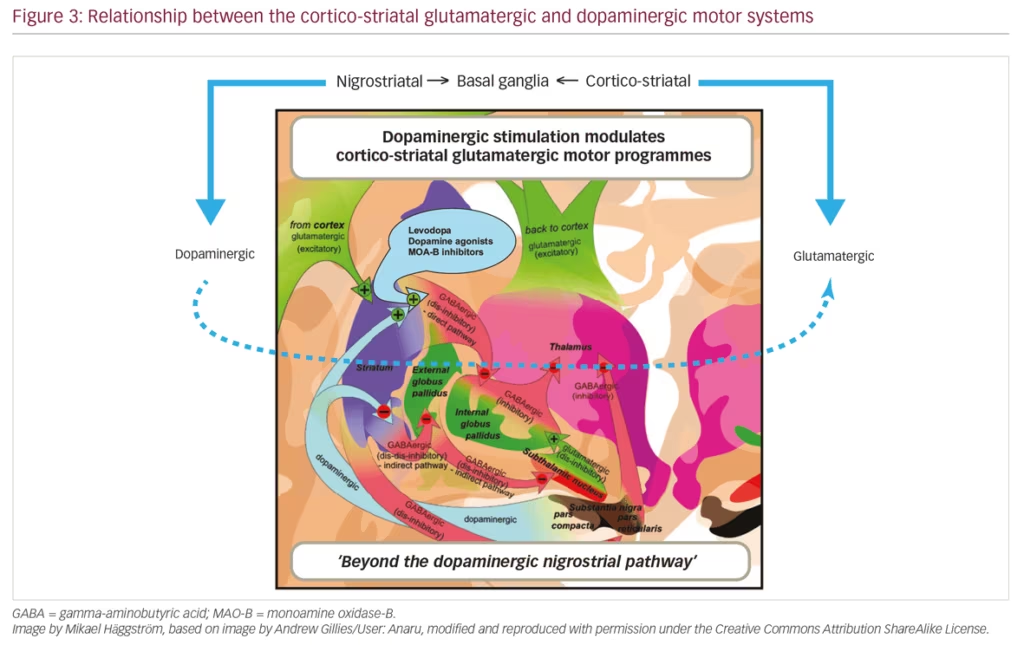
Glutamate plays a pivotal role in normal basal ganglia circuitry. With nigrostriatal dopaminergic depletion, glutamatergic outputs from motor cortical areas and from the subthalamic nucleus to the basal ganglia become overactive, and secondary signalling adaptations at the receptor level are observed in these brain regions. There is also evidence of increased glutamatergic activity within the striatum, affecting mostly the external segment of the globus pallidus in the indirect pathway.24,25 Preclinical models show motor manifestations of parkinsonian symptoms following blockade of glutamate receptors.26 Aberrant glutamatergic signalling is associated with the development of non-motor PD symptoms.2 Preliminary evidence suggests that psychiatric disturbances, such as depression, anxiety, dementia and cognitive changes, are improved following glutamatergic stimulation.2 Altogether, these reports implicate abnormal glutamatergic neurotransmission as a contributor to both the motor and non-motor parkinsonian symptoms.
As observed in other neurodegenerative disease states, deficits in neuronal metabolic dysfunction appear to be significant to the central pathophysiology in PD, in addition to altered glutamatergic neurotransmission. Dopamine cell bodies located in the substantia nigra pars compacta are particularly exposed to oxidative stress and metabolic dysfunction. A reduced capacity to entertain metabolic demands may render these compromised pathways vulnerable to the toxic effects of glutamate, which acts as a neurotoxin in the presence of impaired metabolic cellular function.27–29
Pre- and postsynaptic factors are also likely to influence motor complications observed in patients with PD treated with L-DOPA. Dyskinesias and motor fluctuations are observed more frequently in patients with greater presynaptic denervation.30,31 At the postsynaptic level, patients with greater presynaptic denervation show increased density and sensitisation of striatal glutamatergic receptors (NMDA GluN2A subunit, mGluR5), as well as higher expression of glutamatergic receptors extrasynaptically. All these changes finally cause decreased depotentiation at the striatal level, which is independently related with the development of other motor fluctuations or dyskinesias.32
The involvement of the glutamatergic system in the pathogenesis and symptomatology of PD provides an alternative for exploratory targets for therapeutic intervention in PD. Pharmacologic modulation of glutamatergic hyperexcitability has antiparkinsonian symptomatic effects and may also help to protect the remaining dopamine neurons from excitotoxicity.27 Exploration into drugs that target the glutamatergic system with appropriate pharmacologic selectivity is likely to provide improved treatment strategies for PD.
Adjunct therapy using safinamide
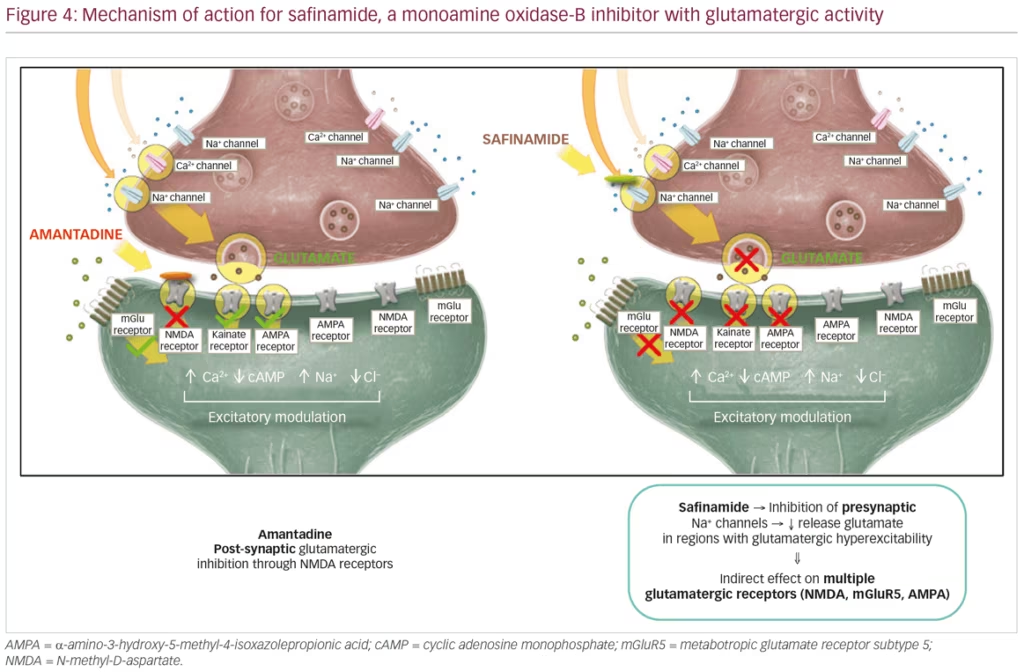
Safinamide (Xadago®, Zambon SpA, Bresso, Italy) is a MAO-B inhibitor and glutamate modulator. Safinamide is approved (EU) for the treatment of mid- to late-stage fluctuating PD as an adjunct treatment to L-DOPA monotherapy or in combination with other PD medications. Safinamide demonstrates two mechanisms of action: 1) safinamide inhibits MAO-B, an enzyme that promotes dopamine degradation, thereby indirectly increasing dopamine levels; and 2) reduces abnormal glutamate release by modulation of sodium- and calcium-ion channels. Specifically, safinamide inhibits presynaptic sodium channels thus reducing glutamate release in neural regions that exhibit glutamatergic hyperexcitability (Figure 4).
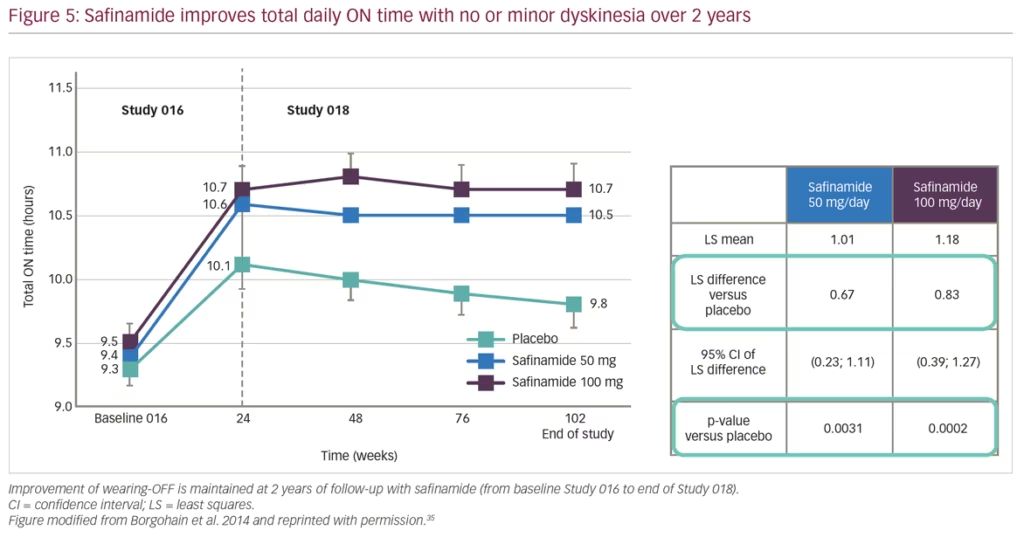
Pivotal studies for safinamide include the phase III, double-blind, placebo-controlled studies ‘SETTLE’33 and ‘016’,34 and its subsequent extension study ‘018’,35 which provided evidence for the long-term efficacy of safinamide as an adjunct therapy to L-DOPA significantly improving motor function. Collectively, evidence from the ‘016’ and ‘SETTLE’ studies found that safinamide improves ON time with no or non-troublesome dyskinesia and motor fluctuations in patients taking L-DOPA only or L-DOPA with other PD medications. Safinamide also showed significant improvement in total daily OFF time, Unified Parkinson’s Disease Rating Scale (UPDRS) part III total score, and Clinical Global Impression-Change score, when compared to placebo.33,34 The subsequent double-blind, long-term extension study 018 provided further evidence for both doses of safinamide (50 mg/day and 100 mg/day) as an adjunct therapy.35 At week 102, a significant improvement in ON time with non-troublesome dyskinesias was observed for both doses of safinamide (Figure 5). In addition, a significant decrease in Dyskinesia Rating Scale (DRS) scores with safinamide 100 mg/day compared with placebo (27% versus 3% respectively; p=0.031) was observed in patients with clinically relevant dyskinesias (DRS >4) at baseline.35
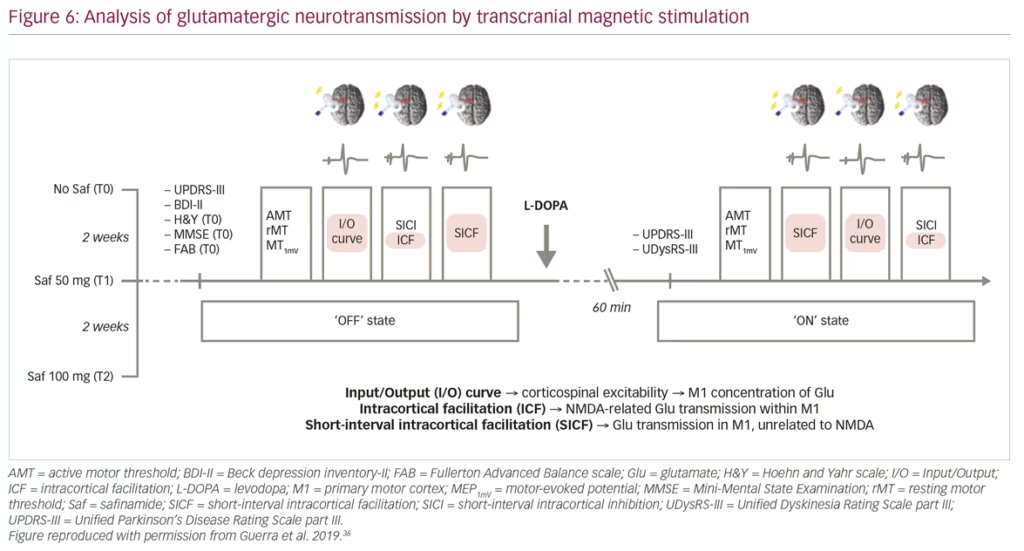
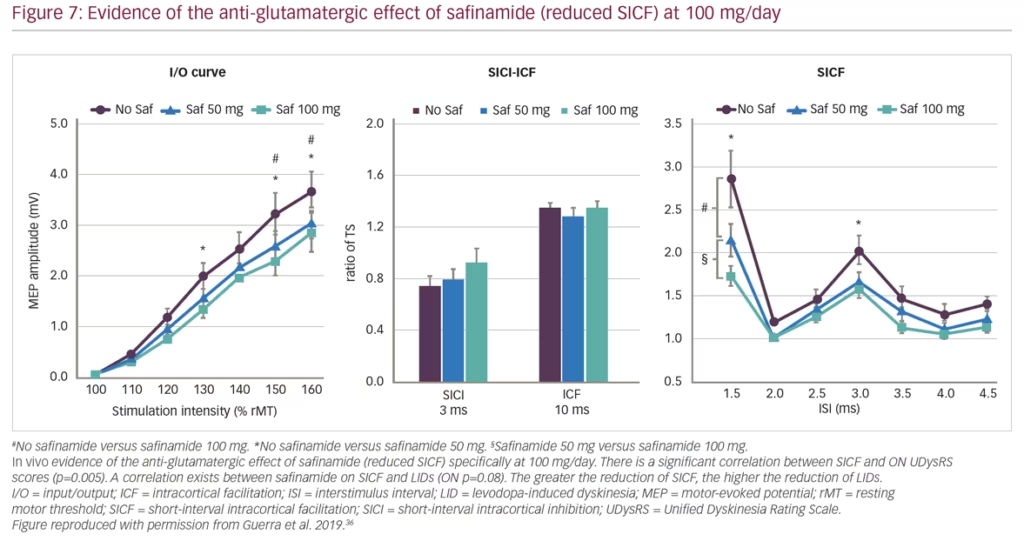
Guerra et al. recently published findings from a study using transcranial magnetic stimulation protocols to assess different facilitatory intracortical circuits in the primary motor cortex of patients with PD that exhibit L-DOPA-induced dyskinesias (Figure 6). The study found that these patients show abnormal cortical facilitation in primary motor cortices, presumed to be due to overactive glutamatergic neural transmission. Safinamide (100 mg/day) was able to restore this dysfunction through anti-glutamatergic properties.36 Transcranial magnetic stimulation mapping correlated reductions in short-interval intracortical facilitation following safinamide with higher reductions in L-DOPA-induced dyskinesias among patients with PD, providing in vivo evidence of the anti-glutamatergic effect of the drug. Subsequent clinical studies have consistently demonstrated improvement in motor function and increased ON time with non-troublesome dyskinesia (particularly at high dose of 100 mg/day) with safinamide as an adjunct therapy combined with L-DOPA (Figure 7).37,38
Additional support for the use of safinamide includes evidence in post-marketing assessments. Mancini et al. observed significant improvements with safinamide treatment in many scales such as the UPDRS part III (motor score), item 9 (walking and balance) of the Unified Dyskinesia Rating Scale, the daily time spent in OFF and ON with disabling dyskinesia, and a reduction of the mean daily dose of concomitant L-DOPA, dopamine agonists and COMT inhibitors.38 The X-TRA study likewise demonstrated improvements in UPDRS part III scores for motor symptoms, Non-motor Symptoms Scale (NMSS), the Abnormal Involuntary Movement Score, and in quality of life (as assessed by the 8-item Parkinson’s Disease Questionnaire [PDQ-8]).39 Additionally, Bianchi et al. showed significant reductions in NMSS (p=0.0003), Clinical Impression of Severity Index (p=0.004) and the PDQ-8 (p=0.0004). Statistically significant improvements in the total score of NMSS was observed and in 6 out of 9 domains, and 13 out of 30 selected items. Improvements in NMSS items include sleep/fatigue, mood/cognition, attention/memory and cardiovascular score, demonstrating the capacity of safinamide to provide non-motor benefits to these patients.40 Therefore, it is our recommendation that, upon the initial development of motor fluctuations, patients with PD be treated early with safinamide as add-on therapy to their current L-DOPA regimen. The addition of safinamide helps to lower total L-DOPA doses and provides further benefit on both motor and non-motor symptoms through its dual dopaminergic and glutamatergic mechanism of action.
Clinical case summaries
Case 1
The patient is a 21-year-old male student who had progressive motor disturbances on the left since age 12. He showed progressive gait disturbances of unknown aetiology despite extensive work-up performed across several medical centres. All diagnostic tests so far were unremarkable including brain and spinal magnetic resonance imaging (MRI) scans. At age 18, he was finally diagnosed with juvenile PD (L>R) related to PARK2 mutations. He had no known family history for PD. [123I]-FP-CIT SPECT imaging showed a severe decrease in striatal tracer uptake, predominant on the right side, which is in accordance with motor symptoms laterality.
The patient described over 95% improvement in motor symptoms following low doses of L-DOPA/benserazide three times a day. Rasagiline, which was started several weeks later, was discontinued because of digestive disturbances. After 9–10 months of low daily doses of L-DOPA, he developed motor complications, characterised by mild wearing-OFF phenomena. Motor fluctuations disappeared after the initiation of the dopamine agonist pramipexole up to 1.05 mg/day and a slight increase in L-DOPA/benserazide to four daily intakes. Over the following months, the patient developed troublesome impulse-control disorders (internet, cell phone, video games), insomnia and suicidal ideas, prompting a rapid discontinuation of pramipexole. Safinamide, 50 mg/day was then started and the patient remained free from impulse-control disorders and troublesome motor complications. Several months later, safinamide was increased to 100 mg/day because the patient reported mild peak-dose dyskinesia. A reduction in the L-DOPA daily dose intervals from 4.0 to 3.5 hours was also recommended because of the recurrence of mild wearing-OFF episodes. At the last visit, the clinical benefit of the association between L-DOPA/benserazide and safinamide persisted: the patient was free from bothersome motor complications with no tolerability problems.
Case 2
The patient is a 59-year-old female hairdresser diagnosed with PD at age 51. There was no known familial history, and the disease was revealed by a resting tremor affecting her right hand. Initial symptoms also included right-sided pain (myalgia and arthralgia), muscle spasms, restless leg syndrome, hyposmia and anxiety. Brain MRI was unremarkable. [123I]-FP-CIT SPECT imaging showed decreased tracer uptake predominant to the left putamen. The patient was successively treated with pramipexole and rasagiline; however, she abruptly discontinued these medications because she had the impression that their overall effect was an increase in muscle spasm and arthralgia. She became reluctant to start other drugs since physiotherapy satisfactorily relieved many of her symptoms.
After 3.5 years, she agreed to start L-DOPA/benserazide, which greatly improved most motor and non-motor symptoms. Slight motor fluctuations were first reported 6 months later, which proceeded to increase in severity 24 months later, in association with painful peak-dose dyskinesia despite divided low L-DOPA/benserazide daily intakes. These bothersome motor complications were greatly reduced following safinamide initiation at 50 mg/day, then 100 mg/day followed by a reduction of L-DOPA/benserazide daily dose. Amantadine was later added to relieve the recurrence of painful dyskinesia. The patient reported significant and prolonged improvement in motor and non-motor symptoms using a combination of L-DOPA/benserazide, safinamide and amantadine.
These cases detail patients who had received a variety of other treatment approaches prior to initiating safinamide. Each treatment approach resulted in the development of unwanted side effects and a non-optimised management plan that did not improve the patients motor status long-term. The use of safinamide from the first fluctuations might have been an alternative approach that could have offered improvement to the patients while avoiding adverse events. It is our recommendation that safinamide be considered as an adjunct treatment to L-DOPA regimens as soon as patients report fluctuations.
Conclusions
- L-DOPA continues to be the gold standard treatment for patients with PD. However, as the disease progresses, response to L-DOPA becomes increasingly short-lived. Both prevention and early management of motor complications is crucial to avoid future development of disabling fluctuations and dyskinesias requiring advanced therapies.
- Adjunct therapies can improve L-DOPA-induced fluctuations. Drugs with multimodal mechanisms of action have shown additional benefits with respect to dopaminergic drugs.
- Rational polytherapy to control dysfunction of dopaminergic and glutamatergic circuits helps to decrease doses of L-DOPA and may modify striatal maladaptive changes.
- Safinamide is an MAO-B inhibitor and glutamate modulator that increases ‘good ON time’ (ON time without troublesome dyskinesias) both in routine clinical treatment and real-life data.
- Real-life data have shown that safinamide also improves non-motor symptoms such as mood/cognition and sleep/fatigue. Also, additional data have shown promising results on pain.
- The differential clinical effect of safinamide on dopaminergic stimulation and glutamatergic modulation is maximised at the dose of 100 mg/day.