Dopamine deficiency resulting from the progressive loss of nigrostriatal dopaminergic cells leads to the motor and non-motor impairments associated with Parkinson’s disease (PD). Treatment strategies are focused on dopaminergic replacement therapy. Levodopa monotherapy may be successful in addressing motor symptoms in early-onset patients and may be optimised by adding on other dopamine-potentiating agents such as monoamine oxidase (MAO) inhibitors, dopamine agonists and catechol-O-methyl transferase (COMT) inhibitors. However, although these strategies can be efficacious in improving motor function, they are also associated with motor complications and fluctuations in ON and OFF time, which are often treatment resistant.1,2
Current understanding of the pathophysiology of PD recognises that neurodegeneration is not confined to the dopaminergic pathway, and in fact there are many nondopaminergic systems that also contribute to Parkinsonian neurodegeneration. In particular, overactive glutamate transmission has been implicated in playing a role in the progression of PD.3,4
Safinamide is a unique molecule with dual mechanisms of action targeting both dopaminergic and non-dopaminergic pathways. As an MAO-B inhibitor, it prevents dopamine degradation. It also provides sodium channel blockade, which leads to the suppression of excessive glutamate release.1 Safinamide has been shown to be an effective adjunct to levodopa therapy, significantly improving motor function and ON time with no/non-troublesome dyskinesia associated with higher-dose dopaminergic intervention.5–7
This review highlights the rationale supporting the adjunctive use of the dopaminergic and non-dopaminergic actions of safinamide as a multifaceted strategy to prolong levodopa efficacy without worsening motor complications. These concepts were initially presented during a symposium at the 21st International Congress of Parkinson’s Disease and Movement Disorders, 6 June 2017, in Vancouver, Canada.
Pathophysiology of Parkinson’s disease beyond dopamine
Although PD is a slow, progressive, degenerative disorder largely characterised by striatal dopaminergic depletion; alterations in other neurochemical pathways, including noradrenergic, serotonergic and cholinergic systems, are also implicated. An imbalance between the dopaminergic and glutamatergic systems is thought to play a particularly critical role in the etiology of PD. Excitatory glutamatergic transmission is increased in dopamine-denervated striatal neurons, which is substantiated by deep brain recordings that have shown higher (abnormal) glutamatergic activity in patients with PD compared with patients with dystonia and tremor. Overactivity of the glutamatergic pathway contributes to motor and non-motor symptoms.8,9
Abnormal glutamatergic transmission in the striatum is considered a key factor in the development of levodopa-induced dyskinesias (LIDs), and indeed alteration of ratios of glutamatergic receptors has been identified as a synaptic trait of LIDs in both experimental models and humans 10
Data from preclinical and clinical studies indicate that activation of glutamatergic receptors, particularly mGlu4, may be effective in anxiety, pain, depression (via α-amino-3-hydroxy-5-methylisoxazole- 4-propionic acid [AMPA]) and sleep dysfunction. Whereas inhibition of glutamatergic neurotransmission may result in increased anxiety and executive dysfunction but can also lessen depression (via N-methyl- D-aspartate [NMDA] receptors) and improve cognitive function.11
PD treatment strategies have tended to focus on dopaminergic replacement therapy. Levodopa monotherapy may be successful to address motor symptoms in early-onset patients and may be optimised by adding on other dopamine-potentiating agents such as MAO inhibitors, dopamine agonists and COMT inhibitors. However, these therapies are not without limitations. Dyskinesia and other motor complications can develop (particularly with levodopa), patients can experience periods of inadequate symptom control, and some PD symptoms can become resistant to treatment over time. Indeed, many non dopaminergic neurotransmitters are now known to be involved in motor symptom control and levodopa-induced motor complications, with overactive glutamate transmission also contributing to PD progression.12
Targeting a pathologic mechanism other than the dopaminergic system is thus emerging as a complementary approach to relieve Parkinsonian symptoms while minimising motor complications and fluctuations.1
Introducing safinamide – a unique dual mechanism of action targeting both dopaminergic and non-dopaminergic systems
Safinamide is a water-soluble, orally active α-aminoamide derivative that modulates dopaminergic and glutamatergic neurotransmission with a unique dual mechanism of action.1
Dopaminergic activity
MAO-B promotes dopamine degradation. Safinamide is a selective and reversible MAO-B inhibitor that prolongs lifetime activity of dopamine in the synaptic cleft (Figure 1).
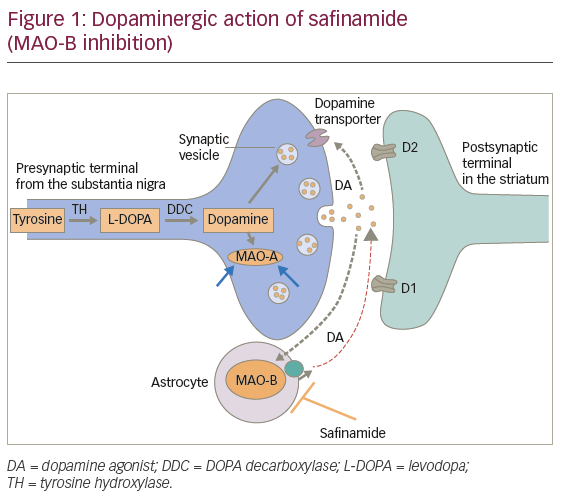
Safinamide is more selective for MAO-B versus MAO-A than selegiline and rasagiline: 1,000-fold in humans versus 127-fold for selegiline and 103-fold for rasagiline. Its reversibility allows for better clinical manageability and limits drug-drug interactions.1
Non-dopaminergic activity
Sodium channels are the primary non-dopaminergic target affected by safinamide. In mid-to-late PD, a profound depolarisation due to persistent sodium channel opening in turn opens voltage-dependent calcium channels, thus allowing the entry of calcium into the synaptic cleft, which triggers the abnormal release of glutamate.1 Safinamide blocks sodium channels in a state- and use-dependent manner, thus modulating calcium channel opening and excessive glutamate release. By inhibiting the abnormal release of glutamate, safinamide may control neuronal hyperexcitability in mid-to-late PD (Figure 2).1

Preclinical data
In preclinical trials, safinamide demonstrated both state-specific and region-specific activity. In rat cortical neurons, safinamide was shown to block sodium channel activity in a state- and use-dependent manner, resulting in depression of neuronal activity at high-frequency firing and ineffectiveness at a normal firing rate.13,14 In monkeys, safinamide maintained increased dopamine levels in the putamen, the region devoted to motor control. It did not affect regions involved in reward circuitry such as the nucleus accumbens, obviating concerns about impulse control.13,14
Based on the preclinical results, it was observed that safinamide reduces striatal glutamatergic overactivity believed to be associated with Parkinsonian motor symptoms. These findings may suggest that the striatal glutamate-modulating component of safinamide’s activity also contributes to its clinical effects, in which long-term use as a levodopa add-on therapy significantly improves motor function and ON time with no/non-troublesome dyskinesia.7
Clinical trials
Safinamide has been evaluated as an add-on therapy to levodopa in two phase III, double-blind, placebo-controlled trials: 016 and SETTLE. In both studies the primary efficacy endpoint was the change from baseline to week 24 in ON time with no/non-troublesome dyskinesia as recorded by patients in an 18-hour Hauser diary. The studies evaluated 1,218 patients with mid- to late-stage PD with motor fluctuations on stable levodopa and other anti-PD drugs.5,6
In study 016, 669 patients were randomised to receive safinamide 50 mg/day (n=223), safinamide 100 mg/day (n=224) or placebo (n=222). At week 24, a significant improvement in ON time with no/nontroublesome dyskinesia versus placebo was noted for both safinamide groups: 50 mg (p=0.0223), 100 mg (p=0.0130). Neither safinamide dose increased troublesome dyskinesia, despite the increased ON time.5
These results persisted out to 2 years in the double-blind long-term extension of this study (018). Overall, 81.3% of patients (544 of 669) previously enrolled in study 016 entered this trial. At week 102, a significant improvement in ON time with no/non-troublesome dyskinesia (a secondary endpoint) versus placebo was noted for both safinamide groups: 50 mg (p=0.0031); 100 mg (p=0.0002).7 The primary endpoint in study 018 – change from baseline (study 016 randomisation) to month 24 in Dyskinesia Rating Scale (DRS) total score during ON time – was not met.7 However, a post-hoc analysis of the subgroup of patients with moderate-to-severe dyskinesia at baseline (36% of patients) showed a significant reduction in DRS score with safinamide 100 mg versus placebo (p=0.0317).7
Results from studies 016 and 018 also show that safinamide used as an add-on to levodopa significantly improves motor function. At week 24, Unified Parkinson’s Disease Rating Scale (UPDRS) III motor examination scores (a secondary endpoint) had improved significantly with safinamide 50 mg and 100 mg but worsened with placebo.5 UPDRS III scores remained significantly lower with safinamide versus placebo at 2 years, indicating that safinamide is associated with a sustained improvement in motor control.7
In the SETTLE study, 549 patients on stable levodopa and other anti-PD drugs were randomised to safinamide (n=274) or placebo (n=275), with the safinamide group starting at 50 mg and increasing per protocol to 100 mg at week 2. A significant difference in ON time was noted in the safinamide group by the end of week 2 (+1.04 hours versus +0.40 for placebo), which was maintained throughout the study. At 6 months, the mean increase in ON time with no/non-troublesome dyskinesia was +1.42 hours compared with +0.57 for placebo (leastsquares mean difference: 0.96 hours; p<0.001).6 The significant benefit over placebo was clinically relevant, allowing patients to achieve improvements of at least 1 hour in motor fluctuations and 30% or more in motor symptoms (Figure 3).6

The safety and tolerability profile for safinamide was favourable. The incidence of treatment emergent adverse events (TEAEs), both newly emergent and re-emergent, was similar between safinamide and placebo groups.5–7 Taken together, the results of these clinical studies confirm the usefulness of safinamide in patients with midto late-stage PD as an add-on therapy to levodopa and other anti- Parkinsonian drugs.1
Post-hoc analysis
In a post-hoc analysis of studies 016/018 and SETTLE, a number of additional advantages for safinamide emerged.15 Safinamide decreased OFF time relative to placebo, irrespective of whether it was being used as the first or subsequent add-on to levodopa (Figure 4).15

Similar efficacy of safinamide was seen in patients with mild (early) and more severe (advanced) motor fluctuations.15 The benefits of safinamide were also reflected in UPDRS scores for activities of daily living and motor function, and individual cardinal motor symptoms of PD (bradykinesia,
rigidity, tremor and gait) during ON time. These results were surprising because the patients were receiving a stable, optimised dopaminergic therapy, so further improvements were unexpected (Table 1).15

Glutamate is involved in depression and pain, in addition to other nonmotor symptoms.8,9 The PRIAMO study, conducted by Barone et al., details the frequency of non-motor symptoms in typical and atypical PD (Figure 5).16

Further post-hoc analysis suggested that safinamide 100 mg/day had a favourable effect on pain, accounting for about 80% of the total effect, as shown by path analysis. The reduction of concomitant pain was about 24% with safinamide versus placebo and was associated with significantly greater improvements in two of the three specific items of the “bodily discomfort” domain of the PDQ-39 questionnaire (Figure 6).17

In another post-hoc analysis, safinamide 100 mg/day improved wellbeing and mood in patients with mid- to late-stage PD. Significantly fewer patients receiving safinamide experienced depression as an adverse event compared with patients receiving placebo, with a positive impact on their quality of life. Mood disorders are a frequent comorbidity in PD patients; the favourable effects of safinamide 100 mg/day on mood may be explained by its modulation of glutamatergic hyperactivity (Table 2).18

Directions for future research
More research is clearly warranted for this promising adjunctive PD therapy. Clinical studies indicate that 100 mg/day may be optimal, but that some patients may still do well on 50 mg/day. Areas of interest for future investigations may also include the effects of safinamide on fatigue, its preventive use for fluctuations in dyskinesia and its effects on cognition in older patients.
Summary and Conclusions
As dopaminergic dysfunction is not the only pathogenic mechanism involved in PD, it is important to target both dopaminergic and nondopaminergic disease pathways. Safinamide, a unique treatment modulating both dopaminergic and glutamatergic systems, has been shown to be effective as an add-on to levodopa therapy, prolonging levodopa efficacy, improving motor function and increasing ON time with no/non-troublesome dyskinesia. Unlike other dopamine replacement therapies that improve motor function by acting solely on the dopaminergic pathway, safinamide does not worsen dyskinesia. This effect may be related to its dual mechanism of action which modulates dopaminergic and glutamatergic pathways. Safinamide is an effective and well- tolerated once-daily drug for fluctuating PD, irrespective of concomitant medication. Post-hoc analyses showed promising benefits in non-motor PD symptoms such as pain and mood. Safinamide has also been investigated in a 24-month study, maintaining its efficacy in the long term. Clinical trials findings to date suggest that safinamide represents an important therapeutic option for mid- to latestage fluctuating PD patients.