Following approval in Europe in 2015 and in the USA in 2017,1 and its subsequent introduction, safinamide has become established as an effective and well-tolerated add-on to levodopa treatment in Parkinson’s disease (PD). Despite the extensive clinical trial data and a growing body of real-world experience of this drug,1,2 there are still aspects of its pharmacology and mode of action that remain to be fully elucidated. Its action as a monoamine oxidase-B (MAO-B) inhibitor is well known, but its action on the glutaminergic and gamma-aminobutyric acid (GABA) systems, and the brain regions that are involved, require further elucidation.1,3 Safinamide is also effective in improving various non-motor symptoms in PD and has been reported to reduce dyskinesia.4 The extent of these effects and how they might change medication regimens in PD need further investigation.
At this point, it is worthwhile reviewing recent preclinical pharmacology studies and how these have changed the understanding of safinamide’s mode of action. It is also of interest to consider how these findings link with data from the pivotal clinical studies and subsequent real-world evidence and what implications this might have on PD management with safinamide. _EPUB-web-resources/image/1.png)
Safinamide – a monoamine oxidase-B inhibitor and a glutamate modulator
Michele Morari
Department of Medical Sciences, University of Ferrara, Ferrara, Italy
The efficacy of safinamide in PD largely results from two different mechanisms of action and it is useful to review recent preclinical evidence demonstrating these different actions and the benefits these bring to clinical treatment.
Safinamide as a monoamine oxidase-B inhibitor
The MAOs are flavoprotein enzymes located on the outer membranes of mitochondria with two main isoforms (MAO-A and MAO-B) that are encoded by two genes on the short arm of chromosome X (Xp11.3_11.4).4–6 These differ in distribution, substrates and inhibitor specificity; both are expressed at certain body sites. MAO-A has been observed in the liver and placenta. MAO-B has been observed in the liver, platelets and lymphocytes. They are also expressed in the central nervous system, including neurons (MAO-A and MAO-B) and glia (MAO-B).7
The roles of MAOs are twofold. Firstly, they deaminate primary, secondary and some tertiary amines including noradrenaline, adrenaline, serotonin, dopamine, phenylethylamine and trace amines.8 Secondly, they detoxify exogenous and endogenous compounds. It is important to note that, in humans, dopamine is metabolised by MAO-B to produce free radicals such as hydrogen peroxide and toxic by-products such as 3,4-dihydroxyphenylacetaldehyde, which can react with cellular proteins including alpha-synuclein and contribute to dopaminergic neuron loss.3,4,9
The MAO inhibitors are long established and successful medications for the palliative treatment of PD, and have been developed through several generations.10,11 The first generation are isoform non-selective and irreversible (e.g., phenelzine and tranylcypromine), the second generation are selective but irreversible (e.g., clorgyline and rasagiline), the third generation are selective and reversible (e.g., moclobemide and reversible MAO-inhibitor antidepressants) and the fourth generation are selective, reversible and multitarget, additionally blocking sodium and calcium channels (e.g., safinamide and zonisamide).12,13 The pharmacodynamic properties of the MAO-B inhibitors have improved with the more recent fourth-generation drug safinamide, showing much shorter offset times and 1,000–5,000-fold selectivity for MAO-B versus MAO-A, compared with 50–250-fold selectivity for selegiline and rasagiline.14–18 In addition, safinamide has substantially greater bioavailability, and a longer Tmax and half-life than selegiline and rasagiline. Both selegiline and rasagiline are metabolised by cytochromes (selegiline by CYP2B6, CYP2A6 and CYP3A4; rasagiline by CYP1A2), making them prone to drug–drug interactions, whereas safinamide has no such interactions.19–21 Selegiline is also metabolised to amphetamine-like metabolites, which may have undesirable effects.22
Safinamide, therefore, has advantages over earlier MAO-B inhibitors in that it is associated with fewer side effects, does not require dietary tyramine restriction, and has a more favourable pharmacokinetic profile.15,22–24
Safinamide as a glutamate modulator
It has now been 20 years since safinamide was first shown to inhibit depolarisation-evoked glutamate release in rat hippocampal slices25 and later in synaptosomes from treatment-naïve rats.15 More recently, acute safinamide treatment (15 mg/kg given intraperitoneally to treatment-naïve rats) was also shown to be effective in reducing glutamate release in the hippocampus, subthalamic nucleus, globus pallidus and substantia nigra reticulata, but not in the dorsolateral striatum.3 In this study, following treatment with a safinamide dose of 15 mg/kg, free brain concentrations of the drug reached 1.89 µM, which are close to the affinity value for the inactivated state of the sodium channel (4.1 µM). This suggested that the inhibition of glutamate release was mediated via the sodium and not the calcium channel.
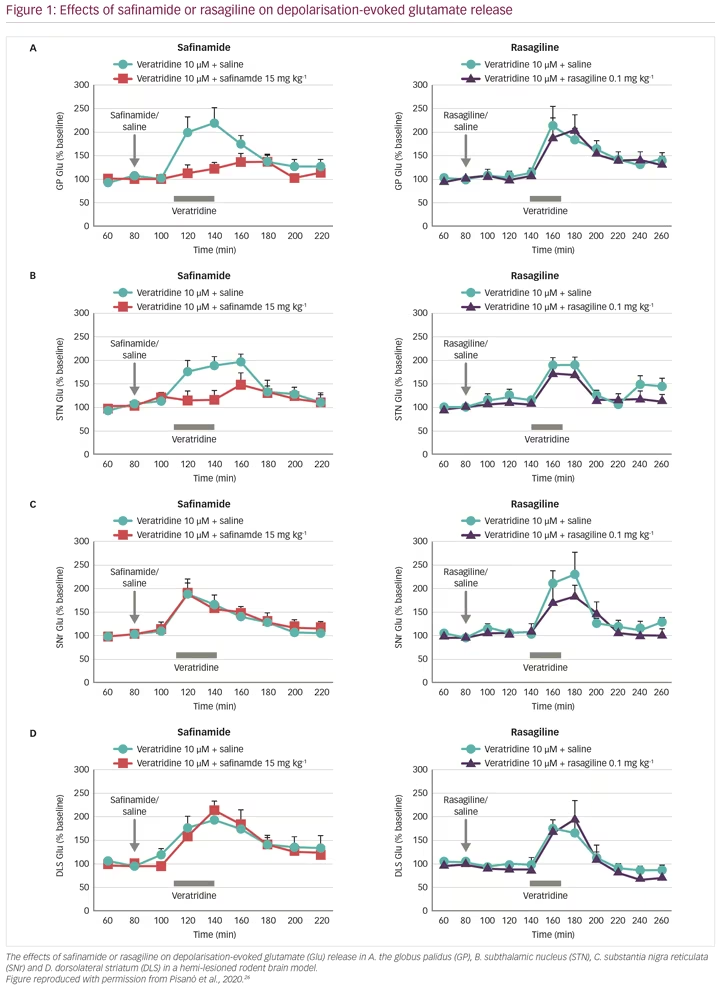
These results raised important questions. In particular: is safinamide also effective in the absence of dopamine? Does MAO-B inhibition contribute to the glutamate-inhibiting effects of safinamide? To address these questions, glutamate and GABA responses were studied in vivo, in the brain of rats where the nigrostriatal dopaminergic pathway was unilaterally lesioned, causing loss of dopamine release, using the parkinsonian toxin 6-hydroxydopamine (6-OHDA).26 The animals were given systemic treatment with either safinamide or rasagiline at doses that would result in >50% MAO-B inhibition (15 mg/kg and 0.1 mg/kg intraperitoneally, respectively), and the brain nuclei under investigation were perfused with veratridine (10 µM for 30 min) through the probe (reverse dialysis) to stimulate glutamate release. In naïve (non-lesioned) rats, safinamide blocked veratidine-induced glutamate release in the globus pallidus, subthalamic nucleus and substantia nigra reticulata, but not in the dorsolateral striatum, whereas veratidine-induced GABA release was unchanged in all these sites (Figure 1, Table 1). In dopamine-depleted rats, however, glutamate release was blocked only in the globus pallidus and subthalamic nucleus, but was unchanged in the substantia nigra reticulata and dorsolateral striatum. In addition, GABA release was blocked in the globus pallidus and substantia nigra reticulata but not in the dorsolateral stratum or the subthalamic nucleus, which was unresponsive to veratridine. In comparison, rasagiline produced no change in glutamate or GABA release at any of these brain sites, indicating that the effect of safinamide is independent of MAO-B.3 These results showed that safinamide effectively inhibits glutamate release in the absence of dopamine in a rodent model. In addition, it could be speculated from the findings that safinamide acts on the subthalamic nucleus/globus pallidus axis.

Evidence supporting the activity of safinamide in the subthalamic nucleus comes from another in vivo PD model study. Rats were given haloperidol, which blocks dopamine D2 receptors in the striatum causing disinhibition of the subthalamic pathway, resulting in akinesia/catalepsy and elevation of glutamate release.27 In these animals, haloperidol raised glutamate release in the substantia nigra, which was reduced by dosing with safinamide.
Additional evidence for safinamide activity at the subthalamic level has been provided by a study on the development of levodopa-induced dyskinesia (LID) in 6-OHDA-lesioned rats.28 The results showed that, despite the inability to attenuate dyskinesia development and expression, treatment with safinamide blocked the rearrangement of the subunit composition of N-methyl-d-aspartate receptors in the striatum (i.e., increased GluN2A/GluN2B ratio in the postsynaptic density of striatal medium-sized spiny neurons), which is a fingerprint of dyskinesia. It also blocked the increase of glutamate release associated with dyskinesia onset in vivo. The results suggested that safinamide modulates cortico-striatal or thalamostriatal glutamatergic transmission, and this may contribute to its clinical benefits. This is particularly apparent in long-term treatment of patients with PD, in which safinamide enhances the motor effects of levodopa without increasing the burden of dyskinesia.
It can be speculated that this chronic effect of safinamide on striatal neurochemistry may be mediated by modulation of the basal ganglia-thalamo-cortico-striatal loop. Indeed, evidence that striatal glutamate transmission can be modulated by the subthalamic nucleus (which is affected by safinamide) has been provided by several in vivo studies. In one example, high frequency stimulation of the subthalamic nucleus in dyskinetic rats was shown to reduce the hyperexcitability of striatal medium-sized spiny neurons caused by 6-OHDA lesions and aggravated by subsequent levodopa treatment.29 Interestingly, persistent sodium channel currents sustain tonic firing discharge of neurons in the subthalamic nucleus.30,31 Additional experiments on rat brain slices showed that high frequency stimulation of the subthalamic nucleus silences glutamatergic subthalamic neurons by inactivation of sodium channel current.32
The inhibition of glutamate and GABA release by safinamide and its suggested action on the cortico-basal ganglia-thalamo-cortical loop is advantageous for a PD treatment, as abnormal discharge of subthalamic nucleus neurons is a key mechanism in the pathology of the motor deficits in the disease.33–37 Correction of this activity improves these symptoms.37–39
The recent animal model findings have expanded knowledge of the mode of action of safinamide in PD. It is known that overactive glutamate transmission inside and outside the basal ganglia is among the factors responsible for motor symptoms, and also for neurodegeneration and non-motor symptoms associated with the disease.5 Therefore, the symptomatic effects of safinamide may be mediated not only by the anti-MAO-B property of the drug, but also result from the normalisation of overactive or abnormal glutamatergic transmission in key areas. This would allow safinamide to extend its symptomatic action over motor symptoms to the control of pain, depression and L-dopamine-induced dyskinesia. The notion that safinamide is capable of normalising abnormal cortical glutamatergic activity through non-dopaminergic mechanisms is further supported by a recent transcranial magnetic stimulation neurophysiological study in patients with PD.40 Here, a 2-week oral treatment of safinamide normalised the enhancement of short-interval intracortical facilitation (SICF), which is an index of cortical glutamatergic neuron hyperactivity, in the primary motor cortex (M1) of patients manifesting levodopa-induced dyskinesia (LID). Interestingly, this effect was found to be independent of MAO-B inhibition.40
In conclusion, the findings obtained in the 6-OHDA hemi-lesioned rats confirm the ability of safinamide to inhibit glutamate release in key motor areas of the basal ganglia under parkinsonian conditions, suggesting this effect contributes to the therapeutic actions of safinamide. Moreover, considering a wider role for the subthalamic nucleus in modulating motor, cognitive and affective functions, these data justify studying the effect of this drug in other models of dysfunction in this brain region beyond PD.
The importance of real-world evidence
Werner Poewe
Department of Neurology, Innsbruck Medical University, Innsbruck, Austria
The evidence base supporting any disease treatment, including medications for PD, can be considered as a hierarchy or pyramid in which filtered information (systematic reviews of randomised, critically appraised topics and/or critically appraised individual articles) is regarded as high quality and sits at the top, followed by individual randomised controlled trials (RCTs). Below this is the unfiltered information (cohort studies, case-controlled studies [case series and reports] and background information and expert opinion), which is regarded as lower strength evidence.41 Real-world evidence is at the bottom of the evidence level pyramid and generally tends to be regarded as lower quality but collectively constitutes an enormous body of data that can provide important pieces of information. Such data comes from varied sources, including pragmatic clinical trials, observational studies, patient registries, claims databases, population health surveys and electronic health records. This type of information is also more likely to be collected during the post-marketing phase, so tends to give a continuing and expanding insight into drug efficacy and safety in the clinical practice at long time intervals after regulatory authorisation. The scale of these sources and time to gain access to collected data varies:42
- prospective studies and hybrid approaches cover >1,000 patients – data access is over 1 year;
- patient-generated data (e.g., social media or patient research networks) cover >100,000 patients – data access is immediate;
- clinical registries cover <2 million patients – data access within 1 year;
- electronic health/medical records cover 2–10 million patients – data access is immediate; and
- administrative claims databases cover >10 million patients – data access is immediate.
In PD, RCTs are regarded as the gold standard and have the advantage of studying matched patient groups. RCTs are conducted along well-defined and tight methodological lines and can be registered to prevent selective reporting. However, drawbacks of such trials are that they are costly and cumbersome to conduct and involve limited numbers of participants that may underrepresent key patient groups.43 In addition, the comparator, such as placebo, is often irrelevant as it does not represent real patient treatment, the study may measure surrogate endpoints rather than actual clinical outcomes, and the protocol may not reflect typical care. Observational trials, however, can involve large numbers of typical patients in routine care settings, they can focus on vulnerable patient groups, can be completed fairly quickly at low cost, can follow patients for many years, can identify rare adverse events and can compare outcomes of treatment alternatives. These trials are limited by confounding factors such as differences among patients receiving different treatments, and differences in patient selection and compliance. They may also indicate outcome associations that are not actually causal.43
Real-world evidence is important and supplements data from RCTs by providing data from large populations, risk–benefit assessment, data on patient groups not previously studied and determine long-term outcomes. This has a potential impact on hypothesis generation, new drug indications and label expansion, and informs treatment guidelines.
Evidence supporting safinamide from pivotal trials and the real world
Heinz Reichmann
Dresden University of Technology, Dresden, Germany
Safinamide has the advantage of acting both on dopaminergic targets, affecting MAO-B, and non-dopaminergic targets, affecting sodium and calcium channels and modulating glutamate release.15 For the latter, safinamide appears to act on only overactive glutamatergic neurones, which is an important selective function in PD treatment. The dual action is currently unique in treatment options for PD, and it raises the question what benefits it provides in clinical use.
Safinamide has been evaluated as a levodopa add-on therapy and as a dopamine agonist add-on therapy for PD in a phase III clinical trials programme that has included >3,000 patients.1 Among these key studies, the trial ‘Safinamide as Add-On Therapy to Levodopa in Mid- to Late-Stage Parkinson’s Disease Fluctuating Patients (SETTLE)’ (n=549) was conducted in Europe, Asia Pacific and North America, in patients who were experiencing motor fluctuations on levodopa therapy, were stabilised on levodopa, and then received either add-on safinamide (50 mg/day for 2 weeks then 100 mg/day) or add-on placebo for 24 weeks.44 The higher dose of safinamide is believed to be necessary to achieve a glutaminergic effect. Patients receiving safinamide showed a slightly greater than 1 hour increase in total daily ‘good’ ON time (ON-time without troublesome dyskinesia) compared with placebo (p<0.001) and a 1-hour reduction in total daily OFF time (p<0.001). The onset of these effects was rapid, becoming apparent after just 1 month. There was also a remarkable least-squares mean reduction versus baseline of 3.43 points in the Unified Parkinson’s Disease Rating Scale (UPDRS) part III score (clinician-scored monitored motor evaluation) for safinamide (p=0.003).
In another key trial, Study 016 (n=669), patients were stabilised on levodopa and then randomised to add-on safinamide 50 mg/day or 100 mg/day or add-on placebo for 6 months.45 This trial was extended by Study 018, in which patients continued to receive randomised treatment for a further 18 months.46 Safinamide add-on treatment resulted in notable and significant improvements in total daily ‘good’ ON time (ON-time without dyskinesia + ON-time with minor dyskinesia) after 6 months, for both doses, over add-on placebo (p=0.0223 and p=0.0130). These improvements were maintained over the following 18 months and were achieved without troublesome dyskinesia; adverse event profiles were similar with safinamide to those of placebo.
In addition to the pivotal clinical trial results, a range of more recent real-world clinical studies have provided valuable insights into the action of safinamide in PD. Animal model studies have shown that increased facilitatory corticostriatal activity reflects overactive glutaminergic neurotransmission that contributes to the pathophysiology of LIDs.40 This was also investigated in the human disease in a recent study of 20 patients and 20 matched controls. This study assessed different facilitatory intracortical circuits in the primary motor cortex (M1) in patients with PD and LIDs using a combination of transcranial magnetic stimulation protocols.40 Prior to treatment, patients with LIDs had shown increased steepness of stimulation intensity versus amplitude input/output curves and this was decreased by levodopa. These patients also showed increased SICF, but this did not decrease with levodopa treatment. However, safinamide at a dose of 50 mg/day decreased the input/output curve steepness and reduced SICF. Increasing the dose to 100 mg/day also restored SICF to normal. In patients with PD and LIDs, the levels of SICF correlated with the severity of dyskinesia. In patients without LIDs, SICF was abnormal to a lesser degree and they were responsive to levodopa. These results indicated that patients with PD and LIDs have abnormal cortical facilitation and potentially have overactive glutamatergic neurotransmission in specific circuits in the M1 motor cortex. The findings also showed that this dysfunction can be rectified by safinamide (100 mg/day) due to its anti-glutamatergic action.
A recent retrospective study examined the efficacy of safinamide 50 mg/day and 100 mg/day, soon after its licensing in Italy, in the treatment of motor fluctuations and dyskinesias in a set of 91 patients with idiopathic PD.47 Between baseline and follow-up after 6 months, safinamide was found to significantly improve PD in terms of UPDRS part III score, United Dyskinesia Rating Scale (UDysRS) walking and balance item 9 and daily time spent OFF and ON with disabling dyskinesias (p<0.0001–0.0008). Safinamide also significantly reduced the mean daily dose of levodopa (p<0.0001), dopamine agonists (p<0.0002), catechol-O-methyl transferase inhibitors (p<0.0039) and levodopa equivalent dose (p<0.0001). This trial was novel in that it was the first to investigate the benefits of switching patient therapy. In patients who had previously been receiving the MAO-B inhibitors rasagiline or selegiline and were switched to safinamide, there were further significant improvements in OFF time (60 min reduced to 30 min, p<0.0001), daily levodopa dose (716.1 mg reduced to 649.1 mg, p=0.0023), and levodopa dose-sparing capacity or mean levodopa equivalent dose (999.8 mg reduced to 857.4 mg, p=0.0039). In patients who had not previously received MAO-B inhibitors, safinamide also produced significant reductions in ON time with dyskinesias (p=0.0016).
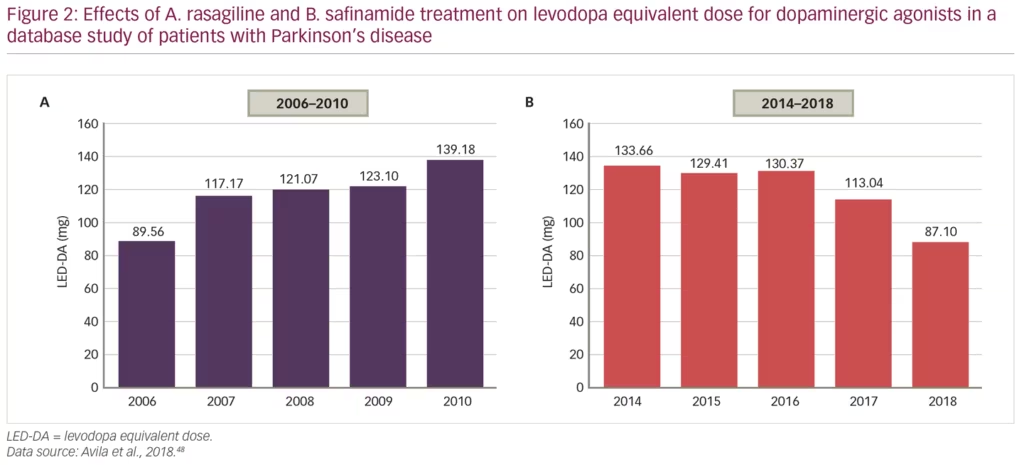
Another recent study analysed records of 724 patients with PD over nearly 20 years, attending a total of 5,124 clinical visits, to compare the capacity of the MAO-B inhibitors, rasagiline and safinamide, to reduce doses of levodopa and/or dopamine agonists, in order to minimise adverse events.48 From 2006–2010, patients mostly received rasagiline and the levodopa equivalent dose for dopaminergic agonists (LED-DA) increased from 90 mg/day to 139 mg/day over that time (p=0.001). From 2014–2018, after safinamide had been started, the LED-DA was reduced over time from 134 mg/day to 87 mg/day (p=0.002) (Figure 2). This improvement could have been affected by other changes in prescription practice over time, but it does suggest that safinamide significantly reduces the mean annual LED-DA and may be associated with reduction in dose-dependent adverse effects in the long-term.
A different aspect of the benefits of safinamide treatment was investigated in a small retrospective study (n=20), in which the effects of safinamide on non-motor symptoms in PD were analysed.4 Data taken from patient questionnaires at baseline and at 4–6 months showed a significant improvement in motor skills (using the SCOPA-Motor Scale; reduction from 24.8 to 14.0 points, p=0.01), but also a numerical improvement in the non-motor symptom scale score, decreasing from 44.3 to 27.2 points (p=0.06). Over the same time period, there were also significant improvements in six non-motor symptom domains: cardiovascular (p=0.02), sleep/fatigue (p=0.001), mood/cognition (p=0.003), attention/memory (p=0.008), urinary (p=0.046) and sexual function (p=0.008). This indicates improved capacity of patients to function in normal life after safinamide treatment.
Motor and non-motor effects were investigated in a further questionnaire-based study that included patients with fluctuating PD who were prescribed add-on safinamide (n=46) or rasagiline (n=15).49 Safinamide significantly reduced both UPDRS part III and UPDRS part IV scores (p=0.01 for both), whereas rasagiline reduced UPDRS part III score (p=0.05) but not UPDRS part IV score. Notably, safinamide significantly reduced items within the Parkinson’s Disease Sleep Scale-2 for items I–V (with the exception of item II), reflecting motor symptoms, dreaming distress, fragmentation of sleep and insomnia symptoms (p<0.05 for all), whereas rasagiline did not significantly reduce any of these items. Safinamide also significantly improved Pittsburgh Sleep Quality Index Component 7, reflecting daytime dysfunction (p<0.05), whereas rasagiline did not. These results appear to indicate that, unlike other MAO-B inhibitors, safinamide may have the additional benefit of improving subjective sleep and daytime somnolence in motor-fluctuating PD.
These findings prompted the initiation of a prospective study (n=30) to further investigate the effect of safinamide add-on therapy (50 mg/day for 2 weeks then 100 mg/day with levodopa alone or combined with other anti-PD drugs) for 6 months on motor symptoms in patients with idiopathic, fluctuating PD. The data are, as yet, incomplete but initial results indicate a positive effect of safinamide on mood, sleep, fatigue and pain.
Overall, the real-world evidence on safinamide use in PD appears to mirror that of clinical trials in that the treatment increases both ‘good’ ON-time and motor functions. Additionally, it has indicated potential benefits on non-motor symptoms such as pain, mood and sleep disorders. Therefore, of all existing add-on therapies available for patients with PD, the evidence suggest that safinamide may currently be the best option for improved control of motor and/or non-motor symptoms. All these benefits appear to be due to the unique properties of the drug having both dopaminergic and non-dopaminergic activity, as well as being fully reversible and highly selective for the MAO-B enzymes.
Conclusion
Safinamide has a well-established efficacy and safety profile in the treatment of PD but there are still aspects of its pharmacology that remain to be fully understood and potentially further exploited. Although its action on MAO-B is well documented, recent in vivo pharmacology studies are beginning to elucidate its role in the striatum to inhibit glutamate release in hyperexcited neurons.3,10,15,28 This is important because it indicates a dual mode of action that includes action on glutaminergic neurons and a potential to decrease or even block LIDs. Further such in vivo studies are warranted to further evaluate striatal, and possibly intracortical, glutamine release inhibition by safinamide and the effects of long-term treatment on these functions.
The pivotal clinical trials that supported the licencing of safinamide as an add-on therapy to levodopa and other therapy in PD provided strong evidence of improvements in motor function in terms of reducing OFF time and increasing the duration and quality of ON times. More recent real-world evidence has provided further strong signals that this advantageous effect, in addition to a levodopa dose-sparing effect, continues in regular clinical use over extended periods. In addition, real-world evidence has provided further signals that safinamide produces improvements in non-motor symptoms, particularly mood, fatigue, daytime somnolence and possibly neuropathic pain. It is also notable that safinamide showed a glutaminergic effect in the brains of patients with PD, which is consistent with in vivo studies. This effect also manifests as a reduction in LIDs in patients receiving the drug. Patients with dyskinesia are often treated with amantadine but the efficacy of safinamide as an alternative treatment or prevention strategy for this symptom requires further evidence.
The pharmacology of safinamide is unique among current PD add-on treatments. In future, the pharmacological and real-world evidence for this drug is likely to encourage its increased use in PD to manage motor and non-motor symptoms and limit the doses and consequent negative effects of long-term levodopa treatment. Greater understanding of safinamide pharmacology and, in particular, its effect on glutaminergic response in patients may also justify its use earlier in the disease course. This change has the potential to limit the emergence of dyskinesia and improve outcomes compared with using levodopa alone or levodopa in combination with other MAO-B inhibitors.