
Alzheimer’s disease (AD), the most prevalent cause of dementia, is a progressive neurodegenerative disorder marked by the abnormal accumulation of amyloid-beta (Aβ) plaques and neurofibrillary tau tangles. It currently affects more than 50 million individuals worldwide, with projections reaching over 210 million by 2050.1 While advancing age remains the dominant risk factor – affecting more than half of individuals older than 85 years – genetic determinants, such as the apolipoprotein E (APOE) ε4 allele and mutations in the amyloid precursor protein (APP) gene, also play pivotal roles in disease susceptibility.2–4 Beyond these non-modifiable factors, modifiable lifestyle and physiological processes are increasingly recognized as crucial determinants of AD risk. Among them, sleep quality and architecture have emerged as particularly influential.5
Emerging evidence suggests that disturbed sleep and AD share a bidirectional relationship: poor sleep promotes amyloid and tau accumulation, while early neurodegenerative changes, in turn, disturb sleep–wake regulation.5,6 This reciprocal cycle may begin years before clinical symptoms appear, providing a potential window for early detection and intervention. Mechanistically, sleep loss is linked to glymphatic dysfunction, impaired Aβ clearance and enhanced neuroinflammation – all of which contribute to neuronal injury and cognitive decline.5,6
Given these converging findings, understanding how sleep disturbances influence AD pathogenesis has important diagnostic and therapeutic implications. This article synthesizes recent experimental and clinical evidence on the mechanisms connecting sleep disruption and AD, emphasizing glymphatic impairment, neuroinflammatory signalling and neurotransmitter imbalance. It also highlights translational opportunities in which sleep restoration or modulation could serve as a preventive or disease-modifying strategy in individuals at risk for AD.
Methods
This narrative review was conducted through a comprehensive assessment of the scientific literature. The databases PubMed, Scopus and Web of Science were systematically searched for studies published between January 2004 and September 2025. Search terms included combinations of: (‘Alzheimer’s disease’ OR ‘dementia’) AND (‘sleep disruption’ OR ‘sleep deprivation’ OR ‘insomnia’ OR ‘slow-wave sleep’) AND (‘amyloid-beta’ OR ‘tau’ OR ‘glymphatic system’ OR ‘neuroinflammation’) AND (‘intervention’ OR ‘therapy’ OR ‘clinical trial’).
The inclusion criteria were restricted to articles published in the English language. Priority was given to studies with higher levels of evidence, including systematic reviews, meta-analyses, longitudinal cohort studies and randomized controlled trials, where available. In addition, studies were considered if they directly examined the relationship between sleep, brain function and the potential for early detection of AD or related neurocognitive outcomes.
Exclusion criteria encompassed non-English publications to ensure uniformity and accessibility of data. The aim was to integrate findings from basic science, translational models and early clinical observations, rather than to perform quantitative synthesis or risk-of-bias assessment. PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) or other systematic review standards were not applied. While this approach enhances conceptual understanding and hypothesis generation, it also introduces the potential for selection bias and limits the reproducibility of search and inclusion processes. These limitations were mitigated by emphasizing peer-reviewed sources and cross-verifying data across multiple high-quality reviews and meta-analyses, where possible.
Basic concepts of sleep
Sleep is broadly categorized into rapid eye movement (REM) and non-REM (NREM) stages, with NREM further divided into light (N1, N2) and deep (N3 or slow-wave sleep [SWS]) stages. SWS is particularly critical for cognitive restoration and the glymphatic clearance of metabolic waste products, including Aβ and tau proteins. Conversely, REM sleep is essential for memory consolidation and emotional processing (Table 1).5–12
Table 1: Sleep disorders and their role in Alzheimer’s disease: Mechanisms, interventions and clinical evidence5–12
| Sleep disorder | Key findings and quantitative evidence | Main interventions/trial results | Proposed mechanisms in AD | References |
| Insomnia | Cohort (n=1,682; 3.5 years): severe insomnia increased dementia risk (HR, 1.53), strongest for AD | CBT-I RCT (n=50, aMCI + insomnia): improved sleep efficiency and memory over 6 months versus control | Reduced SWS impairs glymphatic clearance of Aβ and tau proteins, leading to their accumulation | 7,11 |
| OSA | Multicentre cohort (n=1,639; mean age, 73 years): Each 10 min period of SpO₂ <90% per night was associated with +0.12 Centiloid increase in amyloid PET signal | One-year RCT (n=139, mild-to-moderate AD): CPAP adherence resulted in less cognitive decline on the MMSE and NTB | Hypoxia induces oxidative stress and upregulates BACE1, increasing Aβ production; sleep fragmentation reduces glymphatic flow, enhancing Aβ/tau accumulation | 9 |
| RBD | Longitudinal study (n=1,280 men; 12 years): RBD increased risk of cognitive impairment by ≈2.5-fold (HR, 2.57) | Clonazepam (0.5–2 mg) effective in ≈90% of patients; melatonin (3–12 mg) effective in ≈70–80%; large AD-specific RCTs are lacking | Loss of REM sleep impairs memory consolidation; alpha-synuclein pathology co-aggregates with Aβ and tau, accelerating neurodegeneration | 8,12 |
| Circadian rhythm disruption | Post-mortem analysis (n=189): circadian fragmentation correlated with higher amyloid plaque and neurofibrillary tangle burden and with Braak stage of tau pathology | Four-week RCT (n=70 AD): bright-light therapy increased sleep efficiency by 8.5% and reduced nocturnal wake time by ~30 min | Mislocalization of AQP4 water channels impairs glymphatic clearance, leading to accumulation of metabolic waste products | 5 |
| RLS | Cohort (n=22,192 RLS + matched controls; 3 years): RLS associated with higher AD risk (HR, 1.55), particularly in women and severe cases | Dopaminergic agents (e.g. pramipexole) and alpha-2-delta ligands (e.g. gabapentin) reduce PLMS and improve sleep architecture; no AD-specific RCTs available | Chronic sleep fragmentation and micro-awakenings trigger microglial activation and neuroinflammation, promoting Aβ and tau accumulation | 10 |
AD = Alzheimer’s disease; aMCI = amnestic mild cognitive impairment; AQP4 = aquaporin-4; Aβ = amyloid-beta; BACE1 = β-site amyloid precursor protein cleaving enzyme 1; CBT-I = cognitive behavioural therapy for insomnia; CPAP = continuous positive airway pressure; HR = hazard ratio; MMSE = Mini-Mental State Examination; NTB = Neuropsychological Test Battery; OSA = obstructive sleep apnoea; PET = positron emission tomography; PLMS = periodic limb movements during sleep; RBD = rapid eye movement sleep behaviour disorder; RCT = randomized controlled trial; REM = rapid eye movement; RLS = restless legs syndrome; SpO₂ = saturation of peripheral oxygen; SWS = slow-wave sleep.
Ageing and AD pathology are associated with a disproportionate reduction in SWS and disrupted REM sleep, impairing these vital functions and creating a vicious cycle of neurodegeneration.7 Sleep disturbances extend their impact beyond the brain, correlating with an increased risk of cardiovascular disease, metabolic dysfunction and impaired cognitive performance, including deficits in attention, judgement and hippocampal-dependent synaptic plasticity.8 The process of memory consolidation is highly sleep dependent.13 The hippocampus and associated medial temporal lobe structures are crucial for encoding new information. All information first reaches the hippocampus, which is then classified as important or unimportant. When the information is deemed important, the signal is sent to the mammillary body of the hypothalamus, anterior thalamus, Meynert nucleus basalis of the basal forebrain and memory stores in the neocortex, amygdala and hippocampus.14 Hippocampal information is repeated several times during sleep to create stable long-term memory. N3 is important for the interaction between the hippocampus and neocortex.15 Disruption of this process, as seen with pre-learning sleep deprivation, can impair memory encoding, with studies showing up to a 20% decrease in recall performance.16
Normal ageing effects on sleep and memory
Ageing has multiple effects on the quality and quantity of sleep. These include a reduction in overall sleep duration, REM sleep and N3, but an increase in sleep fragmentation, time spent in light-sleep stages (N1 and N2) and nocturnal awakening.17 This is because, as people age, their circadian rhythms become weaker and less synchronized, and they also encounter more sleep difficulties, including insomnia and sleep apnoea.18
Decreased overnight sleep quality and duration are associated with increased daytime tiredness and naps.19 These changes affect all memory and learning.20 Similarly, the degree of N3 loss is a predictor of poor memory consolidation during night-time sleep. These changes may partially explain the decline in cognitive function observed in old age.21
Although the degree of abnormalities was frequently higher, and there were particular changes in REM sleep, sleep disruptions in patients with AD were comparable to those in healthy older adults, in terms of sleep duration and efficiency (Table 1).22
To improve clarity, a summary table was included to compare different sleep disorders and their respective impacts on AD risk. Evidence suggests that sleep disturbances play a critical role in the pathogenesis of AD by affecting Aβ clearance, tau pathology and neuroinflammation. Table 1 summarizes key sleep disorders, their impact on AD risk and potential interventions.
Pathology of Alzheimer’s disease: possible role of sleep disruption
Sleep disturbance may be crucial to the causative process leading to AD pathogenesis, as well as to fostering AD advancement through a reciprocal, positive feedback loop interaction.
Current research has demonstrated that sleep disruption exacerbates AD pathogenesis through multiple, interconnected mechanisms. The glymphatic system is a brain-wide waste clearance pathway that is most active during SWS, facilitating the removal of soluble metabolites, including Aβ and tau, with APOE ε4 carriers showing amplified risk through compromised aquaporin-4 (AQP4) channel polarization.7,9 Sleep deprivation induces microglial dysfunction, decreasing Aβ phagocytosis by approximately 40% in preclinical models, whereas sleep fragmentation elevates pro-inflammatory cytokines, including interleukin (IL)-6 and tumour necrosis factor-alpha (TNF-α), via nuclear factor-kappa B (NF-κB) activation.23,24 Concurrently, sleep loss triggers endoplasmic reticulum (ER) stress through noradrenergic-mediated activation of the unfolded protein response and disrupts synaptic homeostasis by reducing brain-derived neurotrophic factor (BDNF) expression, while increasing glutamate-mediated excitotoxicity in hippocampal neurons.25,26 These processes create a self-reinforcing cycle, in which neurodegeneration worsens sleep disturbances, which, in turn, accelerates pathological progression. This bidirectional relationship highlights the potential therapeutic value of sleep modulation in AD prevention and management, particularly in high-risk populations such as APOE ε4 carriers.7
Glymphatic clearance and Aβ/tau dynamics
Early signs of memory loss are a hallmark of AD, with brain changes occurring long before clinical signs appear, starting 15–20 years prior to cognitive decline.10 Sleep disturbances, often present in the cellular phase of AD, are key early risk factors for AD development.27 This phase includes the build-up of Aβ plaques and tau tangles, both of which contribute to neuronal damage.28
Aβ plaques, the main marker of AD, arise due to an imbalance between production and removal. Aβ is produced by neurons and removed by the glymphatic system, which is highly active during SWS. This system, which relies on AQP4 water channels, helps to remove waste, such as Aβ and tau proteins.7 Glymphatic clearance efficiency increases by approximately 60% during SWS compared with wakefulness, highlighting the critical role of deep sleep in Aβ clearance.29 Chronic SWS reduction, which is common in ageing, lowers AQP4 channel efficiency, whereas APOE ε4 carriers show exacerbated impairment.7,9 These findings highlight the potential of SWS as a therapeutic target in AD prevention.
While rodent studies clearly demonstrate sleep-dependent glymphatic flux, direct measurement in humans remains challenging.29 Recent neuroimaging techniques provide correlative support, but causal evidence in humans is still emerging, highlighting a key translational challenge.8
Although Aβ has some physiological roles, such as supporting synaptic plasticity and protecting against oxidative stress, its accumulation promotes oxidative damage, further contributing to neurodegeneration.30
It is important to note that while Aβ accumulation is pathogenic, soluble Aβ peptides may play physiological roles in synaptic plasticity and possess neurotrophic properties; the pathological shift likely occurs when clearance mechanisms fail and oligomeric species accumulate.31 Tau protein, which is critical for neuronal stability, interacts with Aβ, exacerbating neurodegeneration through processes such as hyperphosphorylation, which impairs axonal transport and leads to neuronal death.28
Sleep disturbances exacerbate Aβ and tau accumulation. Lack of sleep causes tau and Aβ levels in the cerebrospinal fluid to rise, whereas sleep enables their removal.8 Disruptions in sleep lead to increased protein kinase A activity, which further promotes tau phosphorylation and oxidative stress.32
The orexin system, which is responsible for regulating the sleep–wake cycle, is dysregulated in AD.20 Orexin neurons in the hypothalamus regulate wakefulness, and their overactivity in AD increases Aβ production.33 Dual orexin receptor antagonists (DORAs), like suvorexant, improve sleep quality and reduce Aβ plaques in preclinical models. Clinical trials (High Dose Inorganic Selenium for Preventing Chemotherapy Induced Peripheral Neuropathy [SELENIUM]; ClinicalTrials.gov identifier: NCT04009561) are currently testing DORAs in early AD, offering a potential disease-modifying therapy.20,31,32
Additionally, glymphatic clearance is more efficient during sleep, with increased fluid flow facilitating waste removal, further emphasizing the importance of sleep in maintaining brain health (Figure 1).29
Figure 1: Sleep disturbance and Alzheimer’s disease: A reciprocal relationship and the glymphatic system’s function in amyloid-beta/tau clearance
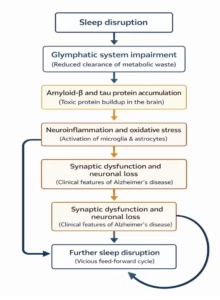
In summary, sleep disturbances contribute to a positive feedback loop, where Aβ and tau accumulation worsen sleep disorders, and disrupted sleep, which, in turn, accelerates the accumulation of these pathological proteins.
Excitotoxicity and apolipoprotein E ε4
APOE plays a role in the regular breakdown of lipoproteins and the transport of fat and fat-soluble substances to the glymphatic system. It is initially synthesized in the brain and the liver. The APOE gene is located on chromosome 19 and has three alleles (ε2=8%, ε3=77% and ε4=15%). APOE ε4 is also associated with AD.34 The pathways of excitotoxicity and APOE ε4 susceptibility do not operate in isolation but rather amplify the core mechanisms already described.35 The excitatory neurotransmitter, glutamate, is responsible for memory and learning. It has two receptors, metabotropic and ionotropic. N-methyl-d-aspartate (NMDA) ionotropic glutamate receptors play a role in AD. Magnesium ion blocks the voltage-dependent calcium channels of NMDA receptors during the resting membrane potential. This barrier is eliminated during depolarization, and calcium influx occurs. These receptors are hyperexcitable in AD because of the reduction in glutamate recycling.10
For instance, sleep disruption-induced glutamate excitotoxicity contributes to calcium overload and mitochondrial dysfunction, thereby exacerbating cellular stress.25 Similarly, the APOE ε4 genotype magnifies the impact of sleep disruption by compounding glymphatic impairment (as discussed in Sleep, Aβ/Tau Clearance and the Glymphatic System) and promoting neuroinflammation.7,9 Thus, these factors are best understood as critical modulators that intensify the primary pathological cycles of impaired clearance, inflammation and neuronal stress.
Endoplasmic reticulum hypothesis
The stress response that causes apoptosis is associated with ER dysfunction. It may be due to either receptor death or mitochondrial dysfunction.36
The pathophysiology of AD includes ER dysfunction. The ER stress pathway is initiated by inositol-requiring kinase 1, which activates kinase 1 signalling and initiates the signalling pathway for c-Jun N-terminal kinase. This cascade activates the pathogenesis of AD through the accumulation of Aβ, phosphorylation of tau proteins and neuroinflammation.37
Sleep disruption increases stress by elevating cerebral noradrenaline levels.38 Norepinephrine acts on α1-adrenergic receptors, causing mitochondrial damage, cytochrome c release and intrinsic apoptosis.25 Therefore, sleep disruption might be involved in AD through the induction of neuronal apoptosis.
Neuroinflammation hypothesis
AD is associated with an increase in inflammatory markers, such as IL-1, IL-6, IL-12, IL-18, interferon and TNF, leading to neuronal cell death and ultimately Aβ and tau protein deposition.11
However, sleep deprivation increases the levels of cyclooxygenase enzymes, TNF-α, IL-1, IL-6 and C-reactive protein.39 Additionally, it triggers the activation of inflammatory signalling factors that can break down the blood–brain barrier, including activator protein 1 and NF-κB. Higher levels of inflammatory markers are linked to shorter sleep durations, which could be the cause of AD.24
Microglia hypothesis
Microglia are immune cells derived from myeloid progenitor cells.40 The risk of AD increases with genetically impaired triggering receptors expressed in myeloid cell 2 genes. Loss of these receptors expressed in myeloid cells exacerbates Aβ plaque-associated toxicity and tau protein toxicity.41
Sleep disturbances, however, affect microglial structure, function and Aβ clearance. Chronic sleep deprivation induces significant morphological changes in microglia, including hyper-ramification and increased soma size, which are associated with enhanced phagocytic activity. These alterations may contribute to synaptic pruning and neuroinflammation observed in early Alzheimer’s pathology.27 Furthermore, studies indicate that sleep fragmentation induces microglial activation, characterized by retracted processes and a transition to an amoeboid morphology, which impairs their ability to clear Aβ. These morphological changes are linked to increased pro-inflammatory cytokine release (e.g. IL-6, TNF-α) and reduced surveillance capacity.40,41
Sleep deprivation reduces microglial Aβ phagocytosis by 40% in mouse models, suggesting a direct pathway linking poor sleep to AD progression.23 Increased microglial phagocytosis associated with sleep disturbances may be exacerbated in patients with AD.
AD may worsen microglial phagocytosis associated with sleep disorders. While these mechanistic insights illuminate the sleep–AD nexus, translating them into clinical practice requires bridging the gap between bench and bedside, a challenge we address in the following section on intervention trials.
Neurotrophic factor hypothesis
Learning, memory, sleep and the longevity of the nervous system depend on BDNF.42 The action of BDNF on tropomyosin receptor kinase B sustains long-term potentiation. Neuronal degeneration and impaired memory formation can result from changes in tropomyosin receptor kinase B receptor levels or BDNF expression.43 Research has revealed that elevated peripheral BDNF levels shield older individuals from AD.42
However, there have been reports of memory and cognitive impairments, reduction in REM sleep and selective alteration of BDNF expression in the hippocampus. The development of AD and cognitive impairment is further exacerbated by sleep disruptions that alter neurotrophic factor levels, which produce reactive oxygen species and neurodegeneration.43
Endocannabinoid system hypothesis
The endocannabinoid system (ECS), involving cannabinoid receptors CB1 (central) and CB2 (peripheral/immune), regulates neuronal excitability, inflammation and sleep–wake cycles. Dysregulation of ECS signalling has been linked to oxidative stress and neuroinflammation – pathways relevant to AD.44 Preclinical models suggest that cannabinoids, such as cannabidiol, can reduce amyloid-β-induced tau hyperphosphorylation and oxidative damage, potentially offering neuroprotection.45 Moreover, activation of CB1 receptors promotes sleep continuity and slow-wave activity, which may indirectly facilitate amyloid clearance.46 However, current evidence remains primarily experimental, and clinical validation of ECS-targeted interventions for AD or sleep disorders is lacking.12 Therefore, while mechanistically plausible, this hypothesis remains speculative and requires further translational research.
Clinical evidence linking sleep disruptions to Alzheimer’s disease
Recent meta-analyses by Irwin et al. and Wang et al. confirm that short sleep duration and poor sleep efficiency significantly increase amyloid deposition and cognitive decline.24,27 However, conflicting findings persist regarding the impact of sleep interventions on biomarker dynamics, underscoring the complexity of translating preclinical mechanisms to clinical outcomes.11 Furthermore, a single night of sleep deprivation can increase tau levels in human cerebrospinal fluid by over 50%.8 Ongoing and recently completed trials now evaluate the clinical and biomarker outcomes of sleep-targeted therapies. Notably, one trial (Trial of Aspirin and Arginine Restriction in Colorectal Cancer; ClinicalTrials.gov identifier: NCT05298721) is assessing cognitive behavioural therapy for insomnia (CBT-I) in individuals with mild cognitive impairment, while NCT04009561 another investigates DORAs in early AD.47,48 Early results indicate that improving SWS may attenuate amyloid accumulation and preserve cognitive performance.
Limitations of current evidence and translational challenges
While the mechanistic links between sleep and AD are compelling, several limitations must be acknowledged, including the over-reliance on preclinical models and limited consideration of individual differences, such as age, sex and genetic background. In clinical settings, adherence to sleep interventions and comorbidities pose additional barriers to translation.17 Human studies, particularly longitudinal cohorts, often rely on self-reported sleep metrics or limited polysomnography, which may not fully capture sleep architecture nuances.
Furthermore, observational studies cannot definitively establish causality due to potential residual confounding. Most clinical trials investigating sleep interventions in AD are still in early phases, and their results are not yet conclusive (e.g. NCT04009561 for DORAs).47 A critical challenge lies in standardizing sleep interventions and identifying the optimal therapeutic window – likely in the preclinical or prodromal stages of AD – for maximum efficacy. Finally, the generalizability of findings may be limited in diverse populations with varying comorbidities and genetic backgrounds. Future research must address these gaps to translate mechanistic insights into effective clinical practice.
Ethical considerations include ensuring informed consent among participants with cognitive impairment, avoiding sedative overuse and balancing intervention benefits against potential risks in vulnerable elderly populations.
Conclusion
In conclusion, compelling evidence supports a bidirectional relationship between sleep disruption and AD, mediated by impaired glymphatic clearance, neuroinflammation and cellular stress. Sleep disturbances are both an early sign and a contributing factor to the disease process. Targeting sleep, particularly SWS, offers a promising avenue for risk reduction. Future efforts must focus on overcoming translational challenges through rigorous clinical trials, as outlined above, to establish sleep health as a cornerstone of AD prevention.
Future research directions
Key unanswered questions must guide future research:
-
Optimal timing and biomarkers: What is the most effective therapeutic window for sleep interventions (preclinical versus clinical AD)? Can sleep architecture parameters serve as reliable, predictive biomarkers for AD risk?
-
Personalized medicine: How do genetic factors, such as APOE ε4 status, modify the response to sleep interventions? Are tailored approaches necessary for different at-risk populations?
-
Mechanistic trials: Well-designed randomized controlled trials are needed to test if improving sleep (via CBT-I, DORAs or SWS enhancement) directly slows the accumulation of AD biomarkers (Aβ, tau) in the human brain.
-
Combination therapies: Could sleep interventions synergize with emerging immunotherapies or lifestyle modifications for greater effect? Promising trials include those targeting orexin signalling and those using multimodal interventions, combining sleep hygiene with other risk-reduction strategies.











