Myasthenia gravis (MG) leads to muscle weakness, which increases with repetitive use of muscles; in the morning, many patients are symptom-free.1,2 Diplopia, ptosis and weakness in other muscles innervated by cranial nerves, are typical. The muscle weakness is, in most patients, generalised with weakness also in the extremities, and sometimes in respiratory muscles. Muscle weakness can be precipitated by repeated muscle use in a test situation.
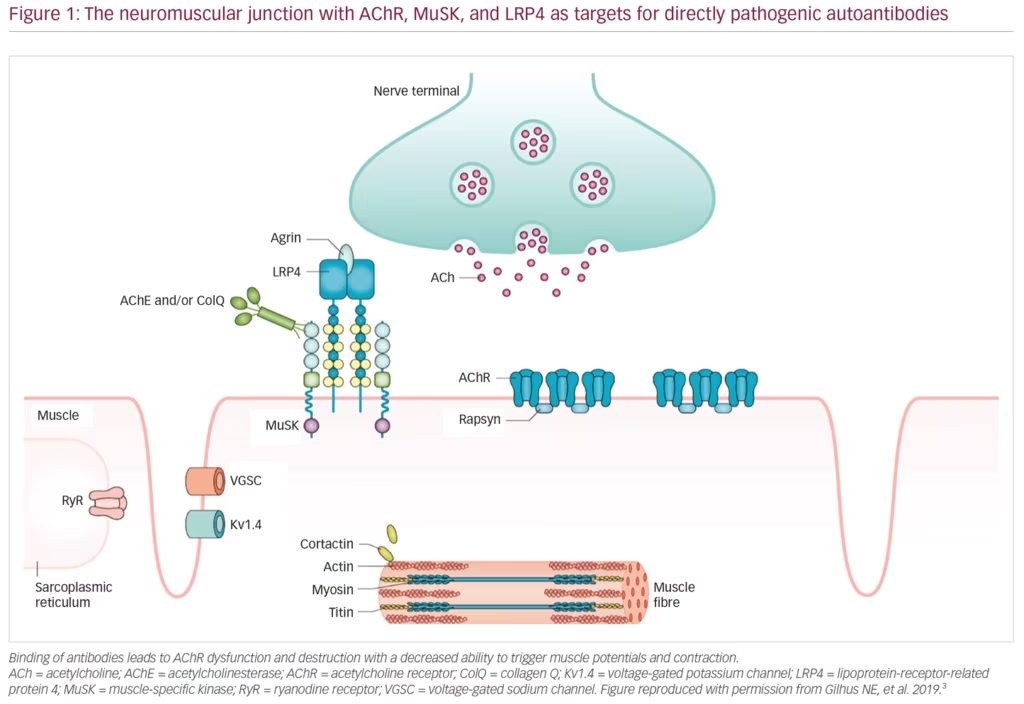
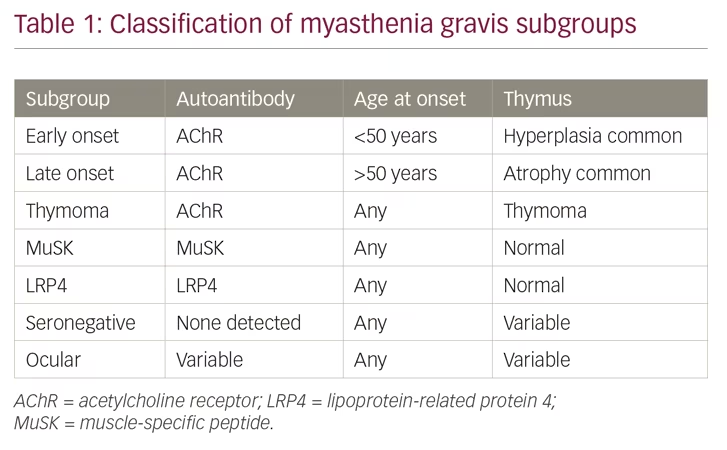
MG is an autoimmune disease caused by autoantibodies that bind to well-defined antigens in the postsynaptic membrane at the neuromuscular junction (Figure 1). Up to 80% of patients have detectable antibodies to the acetylcholine receptor (AChR), whereas 1–15% have antibodies against muscle-specific peptide (MuSK), and a few against lipoprotein-related protein 4 (LRP4).3 Those remaining with no antibodies detected after repeated testing, have either low-specificity/low-concentration antibodies, antibodies against other molecules in the postsynaptic membrane, or a non-antibody-mediated disease. In 10% of patients with MG, the AChR antibody response is induced by a thymoma, and one-third of all patients with thymoma have AChR antibodies.1 A genetic predisposition accounts for approximately one-half of the MG risk, in part, linked to HLA-genes.2 The environmental factors inducing MG have not been identified; however, they most probably differ among MG subgroups. A majority of patients with MG with debut of the disease before age 50 years have thymic hyperplasia with an enlarged gland containing lymphoid follicles. All patients with MG should be categorised into a subgroup due to clinical and biomarker criteria (Table 1).
MG has a prevalence of 150–250 per million in Western populations, with an estimated annual incidence of 8–10 per million.4 Both incidence and prevalence increase with age, but incidence figures show a smaller peak at age 35 years for females.5 The age-adjusted incidence seems to be stable over time. In East Asia, there is a group of early onset MG with AChR antibodies in children <10 years, usually with mild symptoms.6 MG with MuSK antibodies is more common in the southern parts of Europe and in the northern parts of China.7,8 Diagnostic sensitivity and specificity are well above 90% due to antibody testing and neurophysiological examination in clinically suspected cases.
With optimal treatment, the prognosis of MG is generally good.9 Most patients can take part in all daily activities, including work, and their life expectancy is near normal.10,11 However, most patients experience some reduction in their physical capacity, and also in quality of life. About 15% are regarded as relatively resistant to first-line therapies.12 Potential markers for a more severe disease are MuSK antibodies, an increase in antibody concentration during treatment, additional antibodies against the muscle antigens titin and ryanodine receptor, thymoma, and a generalised weakness with involvement of respiratory and bulbar muscles.3 Patients with such biomarkers should be treated more vigorously and long-term.
First-line symptomatic treatment
Acetylcholine esterase inhibition increases muscle strength in MG due to increased availability of the transmitter acetylcholine at the neuromuscular synapse. Pyridostigmine is the favoured drug.13 Ambenonium is an alternative, but is usually less effective, has more variable bioavailability, and is not available in all countries. Most patients manage themselves to find their optimal drug dose, balancing effect on muscle strength and side effects. Dose-limiting side effects include gastrointestinal pain, diarrhoea, increased salivation and other manifestations from the autonomic nervous system. More than 90% of patients with MG with AChR antibodies and with severe disease use a daily acetylcholine esterase inhibitor for the symptomatic effect.9,14 The effect does not diminish over time. Acetylcholine esterases usually have little or no effect in MG with MuSK antibodies, and patients with MuSK MG may even be intolerant to such drugs.7 Pyridostigmine is regarded as safe during pregnancy.15,16
First-line immunosuppressive treatment
Patients with severe MG need to start immunosuppressive therapy when they have been diagnosed; symptomatic therapy alone is not enough (Table 2). The combination of prednisolone and azathioprine is recommended as the first-line treatment in most guidelines and consensus reports.1,13,17 Prednisolone usually induces an improvement after a few weeks, whereas azathioprine often takes several months to improve the condition. Prednisolone has dose-dependent side effects that are well known and the drug is often given on alternate mornings, as this regime may reduce these side effects while keeping the therapeutic effect. Long-term therapy over years is needed in severe MG, often life-long, but the dose of prednisolone can often be reduced to ≤10 mg per day. Both prednisolone and azathioprine are regarded as safe during pregnancy.15,16 Ten percent of patients with MG are intolerant to azathioprine, in part due to variations in the enzyme thiopurine S-methyltransferase.1 Enzyme activity can be measured before initiating the drug. Blood cell counts and liver function should be monitored regularly during the first 2 months of azathioprine therapy.
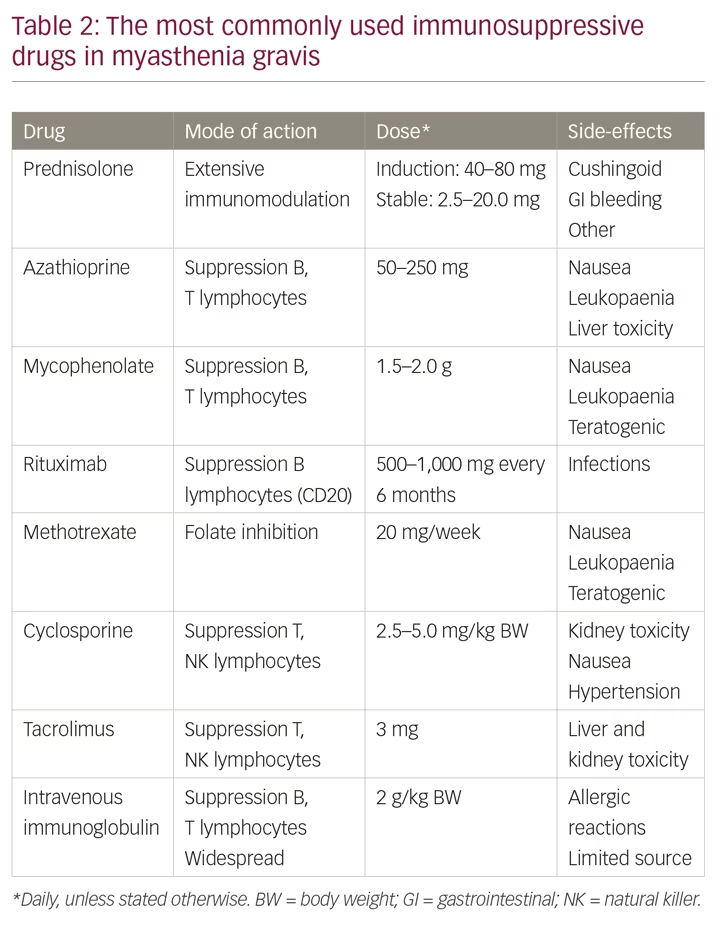
Mycophenolate mofetil is an alternative to azathioprine as a first-line immunosuppressive treatment. Controlled and prospective studies gave disappointing results,18,19 but long-term, real-life therapy seems to have given a better outcome for patients with severe MG. Mycophenolate is teratogenic and should therefore be avoided in all women with the potential of becoming pregnant.
Intravenous immunoglobulin, or alternatively plasma exchange, can be given to patients with severe MG at the time of diagnosis. More than 70% will improve markedly during the first week of such treatment.20 Thus, a remission can be induced, a remission that should be maintained through continuous and long-term immunosuppressive drug therapy. Intravenous immunoglobulin and plasma exchange need hospitalisation and represent relatively expensive therapy, but are safe and generally regarded as cost-effective in exacerbations of severe MG.
Total thymectomy represents first-line therapy in nearly all patients with a thymoma. It is necessary to remove the tumour to avoid local infiltration in the mediastinum, heart and lungs. Small thymomas in elderly and fragile patients with MG may be observed with regular computed tomography (CT) or magnetic resonance imaging (MRI) examinations of the mediastinum. Thymectomy should also be undertaken in all patients with early onset MG with AChR antibodies.21 The age limits for thymectomy have been discussed.3,21 Thymectomy is now widely undertaken up to age 65 years at most centres and is also safe in children >5 years. The positive long-term effect of thymectomy with ocular symptoms only has not been firmly established.22 Some patients without detectable AChR antibodies may benefit from thymectomy as they have low-affinity or low-concentration AChR antibodies not detected in routine assays.23 However, such patients are difficult to identify in a clinical setting. Patients with MuSK antibodies have no involvement of thymus and no effect of thymectomy. During thymectomy it is necessary to remove all thymus tissue, also satellite tissue embedded in mediastinal fat. Video-assisted thymectomy with or without use of robotics is expected to give the same long-term results as open surgery with sternum split. Less invasive surgery is preferred in most centres and leads to a faster immediate recovery.
Second-line immunosuppressive therapy
If there is an insufficient clinical effect of first-line therapies, or in the case of significant side effects, one should initiate alternative immunosuppressive drugs, which should lead to minimal or moderate improvements in MG symptoms only. Our treatment attitude for patients with MG should be proactive and ambitious. Rituximab represents a B-cell directed therapy and should, therefore, be well suited for antibody-mediated diseases such as MG. Many uncontrolled case-based series have reported excellent effect in up to 70% of patients with severe MG.24,25 However, the only placebo-controlled, prospective and blinded study confirmed that rituximab was safe in MG, but failed to demonstrate clinical benefit (ClinicalTrials.gov Identifier: NCT02110706). In a series of 38 patients with MG treated in a single centre for severe and often refractory MG, followed for a mean of 55 months, 28 patients improved and 10 of them with a remission.26 Similar improvements were recently reported in a series of 12 patients with refractory generalised MG from China.27 Rituximab is recommended as a second-line immunosuppressive drug in severe MG and it is especially effective in MuSK MG.7 Long-term therapy is necessary in most patients in combination with the first-line drugs. Alternative second-line immunosuppressive drugs are methotrexate, cyclosporine and tacrolimus.1,2,13 For all immunosuppressive drugs, it is necessary to control and assess the amount of general immunosuppression and take into consideration the slightly increased risk for infections. Dosing of the drugs should depend on the clinical situation. MG manifestations may vary over time, leading to adjustment of drug doses.
Intravenous immunoglobulin improves severe MG rapidly and the effect lasts for 3–4 months. Some patients receive such treatment with regular intervals and over time to maintain a stable improvement. Subcutaneous administration of immunoglobulin is possible and is more convenient for most patients.28 However, the therapeutic effect is, to date, less well documented than for the intravenous treatment.
Third-line immunosuppressive therapy
The C6 complement inhibitor eculizumab represents a new and effective treatment for MG that appears to be well tolerated. In most patients, the effect is moderate, but some patients improve more than with other drugs.29 Long-term treatment seems to be necessary to maintain the improvement. Exceptionally high drug costs mean that eculizumab, in the great majority of patients with severe MG, does not fulfil cost–benefit criteria.30 Alternative complement inhibitors are under way.31 Complement inhibition should be a useful therapy for a disease where complement-binding antibodies mediate cell-membrane destruction and muscle weakness.
Autologous haematopoietic stem-cell therapy is a promising therapeutic option in severe and refractory MG.32 Reported single cases and small case series have shown remission in several patients. At least seven such patients have been reported, with a durable, complete, stable remission and without any other MG therapy.3 No controlled studies have been published, and indications for stem-cell therapy in MG have not been defined. Both cost–benefit and risk–benefit considerations should be important.
Acute exacerbations
Intravenous immunoglobulin and plasma exchange are equally effective for MG exacerbations. Both are fast-acting, with a marked effect after just a few days.20,33 Thus, these treatments represent cornerstones in MG treatment and can be life-saving during severe exacerbations. Megadoses of corticosteroids can be added in patients with an insufficient response. Although intravenous immunoglobulin and plasma exchange have the same response rate of around 70%, individual patients may respond to one of the treatments but not to the other. Intravenous immunoglobulin or plasma exchange can be given before expected exacerbations, for example before major surgery. Plasma exchange probably has a slightly higher risk for severe complications compared to intravenous immunoglobulin.
The threshold for intensive care observation is low during MG exacerbations. Respiratory muscle strength may deteriorate rapidly with the need of assisted ventilation. Aspiration is also a threat. Infection is the most common precipitation factor for acute MG exacerbations. Therefore, infections should be treated vigorously in MG. The antibiotics telithromycin (in particular), fluoroquinolones, macrolides and aminoglycosides should be used with caution as they can induce an MG deterioration.3
Supportive therapy
A well-adapted training programme can improve muscle strength and general well-being in patients with severe MG,34,35 and all patients should be encouraged to take part in such training. Being overweight will reduce physical capacity and should be avoided. Ptosis and double vision can be counteracted with local devices.22 Comorbidity often represents a challenge, especially in older patients, which should be considered when judging the various aspects of the patient’s weakness, mix of symptoms, and reduced health, as well as when trying to combine various treatments for an optimal result. Cardiovascular disease, lung disease and diabetes are all common comorbidities. Patients with MG have a markedly increased prevalence of all autoimmune disorders.36 Repeated antibody concentration measurements (AChR, MuSK) may be helpful in deciding if a deterioration in health is due to worsening of MG or to another medical condition.37 If the expected response to therapies is not achieved, the diagnosis of all patients with MG should be re-evaluated. This is especially important in patients without antibodies and in patients with established or suspected comorbidities.
Myasthenia gravis in children and during pregnancy
Among children with MG, only a few have a severe disease.6,38 Children with MG usually respond well to thymectomy, first-line immunosuppressive therapy and symptomatic therapy. The possibility of growth inhibition as a prednisolone side-effect should be considered. Complete stable remission and pharmacological remission are more common in childhood MG than later in life.
MG is usually unaffected by pregnancy.15 An increased weakness in the post-partum phase is common. Mycophenolate and methotrexate are teratogenic and should not be given at time of conception and in pregnancy.16 Pyridostigmine, azathioprine and corticosteroids are regarded as safe. Ten percent of the babies have a transient, and usually mild, neonatal myasthenia.39,40 In rare cases, the AChR antibodies that cross the placenta bind so severely to the babies’ postsynaptic membrane that they induce malformations, permanent muscle weakness or spontaneous abortion.41 This outcome can be prevented by treatment with intravenous immunoglobulin or plasma exchange during the pregnancy. This should be done in patients with a previous history of such an outcome. Most patients with MG should give birth without any specific precautions. Caesarean section is slightly more common for the MG group.
New treatments
Patients with severe MG need better therapy. Most patients have a reduced quality of life, have symptoms of weakness, and side effects of their current therapy are common. Most patients with severe disease do not reach a complete stable remission or complete pharmacological remission.9 MG cannot be prevented.
Complement inhibitors are already on the market but are still too expensive for ordinary use. FcRn antagonists have shown promising results in phase II studies. Efgartigimod was safe and well tolerated in 35 patients with MG.42 Seventy-five percent of the patients had a rapid and clinically meaningful improvement. Phase III studies are now ongoing, and new B-cell inhibitors, in addition to rituximab, are on trial.3 Beneficial effects have been reported in single patients on the anti-interleukin-6 drug tocilizumab,43 and the JAK1 and JAK2 inhibitor, ruxolitinib, has also improved a patient with MuSK MG.44 Many other new immunomodulatory drugs would be expected to have the potential to improve MG, but studies are lacking for a disease that is relatively rare, and which, in most patients, is well-controlled on standard therapy.45 Specific therapies for AChR and MuSK antibodies have been developed in experimental models, but are not yet in use for human MG.3,45 Experimental MG in rats could be prevented by vaccination with AChR cytoplasmic domains, and established experimental MG was reversed in the rats.46
Conclusions
Most patients with severe MG do well. They are able to take part in ordinary daily life and they do not have a reduction in life expectancy. However, even with optimal therapy, they have reduced muscle strength and physical capacity, side-effects from their symptomatic and immunosuppressive therapy, and a reduced quality of life. There is a need for new therapies with even better treatment effects and fewer side effects, while fulfilling cost–benefit aspects. The aim for all patients with severe MG should be complete stable pharmacological remission or minimal symptoms only.