Overview of topics covered in this review
Glutamate and the basal ganglia in the healthy brain
Glutamate and glutamate receptors – role in motor circuitry
The basal ganglia – anatomy and function
The basal ganglia pathways and the role of glutamate in the healthy brain
Glutamate and the basal ganglia in the parkinsonian brain
Glutamate and Parkinson’s disease pathogenesis
Glutamate excitotoxicity and Parkinson’s disease progression
Clinical implications of glutamate overactivity in Parkinson’s disease
Motor symptoms
Motor complications and levodopa-induced dyskinesia
Non-motor symptoms
Glutamate as a potential target for Parkinson’s disease pharmacotherapy
Modulation of glutamate receptors
Inhibition of glutamate release
Modulation of glutamate uptake
Parkinson’s disease (PD) is a central nervous system (CNS) degenerative disorder that is associated with typical motor symptoms, notably hypokinesia, bradykinesia, rigidity and resting tremor. PD is characterised by a striatal deficiency of the neurotransmitter dopamine, which is due to a progressive loss of nigral dopaminergic neurons. In addition, glutamatergic neurotransmission – the major excitatory system in the brain – and other neurotransmitter systems, such as inhibitory gamma-aminobutyric acid (GABA)ergic neurotransmission, play a critical role in the pathophysiology of PD.
First, this review addresses the role of glutamate in the basal ganglia in the context of a healthy brain, describing in particular motor circuitries. Following this, the pathophysiology of glutamate overactivity in PD is considered, along with the resulting motor and non-motor symptoms, before moving on to explore glutamatergic circuitry as a target for pharmacotherapies in PD.
Glutamate and the basal ganglia in the healthy brain
Glutamate and glutamate receptors – role in motor circuitry
Glutamate is the principal excitatory neurotransmitter in the CNS. It plays a central role in fundamental brain functions, including synaptic plasticity (important for learning and memory), formation of neural networks during development and repair of the CNS.1,2 Glutamate is also essential in the control of movement, due to its actions in neural circuits of the basal ganglia.3 Under certain conditions, however, glutamate can damage nervous tissue and is implicated in several brain disorders, including PD.4
Glutamate synthesis, release and metabolism
Glutamate is a non-essential amino acid with restricted passage to the brain from the blood. In the CNS, glutamate is synthesised in neurons as part of the glutamate–glutamine cycle.5,6
1. Glutamine, the most prevalent precursor of glutamate, is released from neighbouring glial cells and taken up by neuronal presynaptic terminals via excitatory amino acid transporters (EAATs).
2. Within the presynaptic terminals, glutamine is converted to glutamate by the mitochondrial enzyme glutaminase.
3. After its synthesis, glutamate is packaged into synaptic vesicles by vesicular glutamate transporters.
4. During neurotransmission, glutamate is released from the vesicles into the synaptic cleft, where it interacts with receptors that are located on the postsynaptic neuron. The release of glutamate from the presynaptic neuron is triggered by an action potential, as follows:
- an action potential causes voltage-gated ion channels to open in the cell membrane of the presynaptic neuron, allowing cations to enter or exit the cell;
- rapid influx of the cation Na+ alters the membrane potential of the neuron. The resulting depolarisation propagates the action potential further along the length of the neuron. This depolarisation also causes voltage-gated Ca2+ channels to open, allowing the influx of Ca2+; and
- at the presynaptic terminal, this Ca2+ influx triggers glutamate vesicles to fuse with the cell membrane and release glutamate into the synaptic cleft.
5. Glutamate is removed from the synaptic cleft by EAATs, which transport glutamate into glial cells or back into the presynaptic terminal. Glutamate is present in high concentrations in the synaptic cleft for only a short period.
6. In glial cells, glutamate is converted back to glutamine by the enzyme, glutamine synthetase.
Thus, neurons and glial cells work together, synthesising and recycling glutamate to ensure that an adequate supply is available for neurotransmission.7
Glutamate receptors
As described above, glutamate is released from the presynaptic neuron and interacts with its receptors on the cell membrane of the postsynaptic neuron. There are several types of glutamate receptor that, when triggered by glutamate, work together to modulate excitatory postsynaptic neurotransmission.8The specific receptors that are activated by glutamate can be classified into two major families: ionotropic glutamate receptors and metabotropic glutamate receptors (mGluRs).8 Ionotropic glutamate receptors are ligand-gated ion channels and include the NMDA (N-methyl-d-aspartate), AMPA (a-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) and kainate receptors, all of which share a similar structure but differ in their amino acid sequences, subunit combination, and agonist sensitivity/selectivity. mGluRs belong to the G-protein-coupled receptor family and are classified into three groups (I, II and III) according to sequence similarity, signal transduction mechanism, and pharmacological properties.8,9
Ionotropic receptors
Whereas the opening of voltage-gated ion channels is dependent on membrane potential, ligand-gated ion channels require the binding of a ligand, such as glutamate. When glutamate binds to an AMPA or kainate receptor on the postsynaptic neuron, the ligand-gated ion channel opens, allowing a rapid influx of Na+.10 This temporarily depolarises the cell membrane, producing an excitatory postsynaptic response which can initiate an action potential. AMPA receptors mediate most of the fast excitatory neurotransmission in the brain, whereas kainate receptors are thought to have a modulatory role.
In contrast, the opening of NMDA receptor ion channels requires three events to occur: 11,12
- the binding of glutamate;
- the binding of a co-transmitter at a different site (commonly, the amino acids glycine or d-serine); and
- depolarisation of the cell membrane. At rest, NMDA receptors are blocked by a magnesium ion (Mg2+). Depolarisation, which can arise from activation of AMPA/kainate receptors as described above, is required to relieve the NMDA receptor of its Mg2+ blockade.
Upon opening, the NMDA receptors allow the entry of Ca2+, in addition to Na+. Ca2+ is thought to act as a second messenger, activating the intracellular signalling cascades that are associated with long-term potentiation and synaptic plasticity – the major cellular mechanisms that underlie learning and memory.13
Ionotropic receptors are present in all nuclei of the basal ganglia, with higher density in striatum.
Metabotropic receptors
Metabotropic receptors are not ion channels; instead, the binding of glutamate to a metabotropic receptor activates a G-protein within the cell.14 G-proteins act as second messengers, activating intracellular signalling cascades; via these pathways, the activation of mGluRs indirectly modulates postsynaptic ion channels.15
Metabotropic receptors are widely distributed in all nuclei of the basal ganglia16 and are associated with a slower postsynaptic response than ionotropic receptors; their stimulation can result in either increased or decreased excitability. Group I mGluRs are expressed on postsynaptic membranes,17,18 where they are thought to facilitate and strengthen responses mediated by ionotropic receptors. In contrast, group II and group III mGluRs are mainly expressed on presynaptic membranes, where they may function as autoreceptors, providing feedback to downregulate glutamate release.19,20
The basal ganglia – anatomy and function
In order to discuss the significance of glutamate in PD, mechanisms of motor control in healthy individuals must first be considered. The basal ganglia are a group of nuclei found deep in the cerebrum that play a vital role in controlling movement. The nuclei comprise the caudate nucleus and putamen (together forming the corpus striatum), the globus pallidus, the substantia nigra (in the midbrain), and the subthalamic nucleus (located just anterior to the substantia nigra) (Figure 1).21 Together, the basal ganglia receive and process sensory and motor information to coordinate voluntary motor activity.
Figure 1: Motor components of the human basal ganglia and associated structures
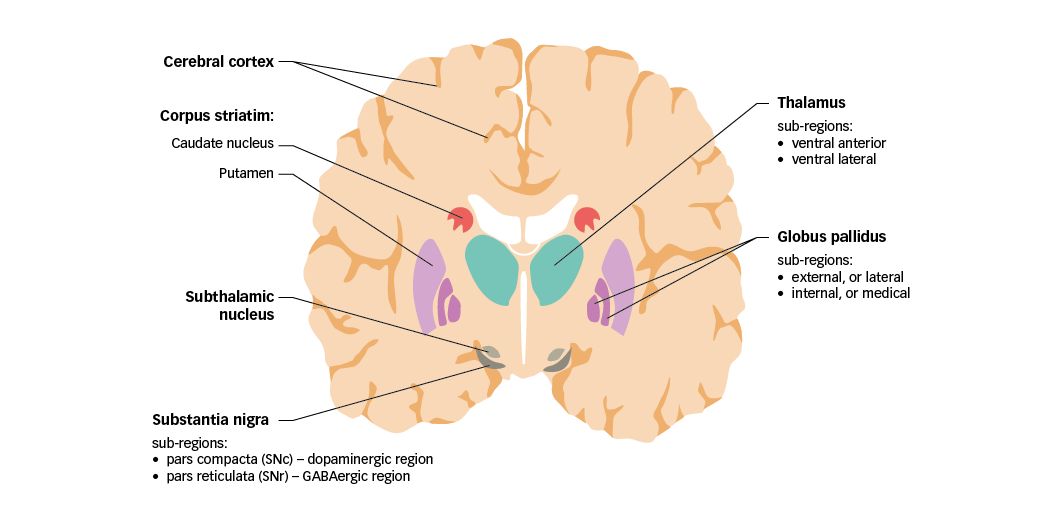
GABA =
gamma-aminobutyric acid; SNc = substantia nigra pars compacta; SNr = substantia nigra pars reticulata. Adapted from Purves et al.5
Sensory and motor input to the basal ganglia is received from specific regions of the cerebral cortex, with substantial contributions from the frontal, parietal, and temporal lobes. Nerve projections from these regions are mainly received by the corpus striatum, and are therefore referred to as the cortico-striatal pathway. Of note, cortical inputs to the caudate nucleus and putamen arise from functionally different regions, implying that the cortico-striatal pathway comprises parallel, interacting pathways with specialised functions (sensorimotor, cognitive and emotional–motivational).22 The basal ganglia also receive input from certain thalamic nuclei, and from the substantia nigra pars compacta (SNc; discussed in the following section).
The main sources of output from the basal ganglia circuitry are the internal globus pallidus (GPi) and the substantia nigra pars reticulata (SNr). Together, these two regions are referred to as the basal ganglia output nuclei. Signals emanating from the basal ganglia output nuclei ultimately target premotor neurons in the frontal cortex and brainstem. Neurons in the GPi project their axons mainly to the ventral anterior (VA) and ventral lateral (VL) nuclei of the thalamus, which in turn project to motor areas in the frontal cortex. These pallido-thalamic projections are particularly important for the control of limb movements. The SNr projects to the thalamus, but also to several premotor nuclei in the brainstem. Notably, SNr projections to the superior colliculus are important to co-ordinate eye and head movements.23
A distinctive feature of basal ganglia organisation is that all efferent nerve projections (except for those originating in the subthalamic nucleus and SNc) use the inhibitory neurotransmitter GABA as their main signalling molecule. Glutamate, however, is vital to regulate information processing in the basal ganglia, being the neurotransmitter used by both cortico-striatal and thalamo-cortical projections, and by efferent nerve projections originating in the subthalamic nucleus.24
The following section further considers the connections between nuclei of the basal ganglia, and expands on the role of glutamate in this circuitry.
The basal ganglia pathways and the role of glutamate in the healthy brain
Connections between the nuclei of the basal ganglia are complex and their physiological implications are not fully understood. However, there is consensus around the existence of two functional pathways linking the striatum with the basal ganglia output nuclei – the ‘direct’ pathway and the ‘indirect’ pathway, which together regulate motor activity (Figure 2).25
Functional interactions between the direct and indirect pathways continue to be a topic of intense research.26–29 Nevertheless, the simplified model presented is well-accepted and provides a framework to consider the changes occurring in the PD brain.
Figure 2: Basal ganglia functional motor circuitry – direct and indirect pathways
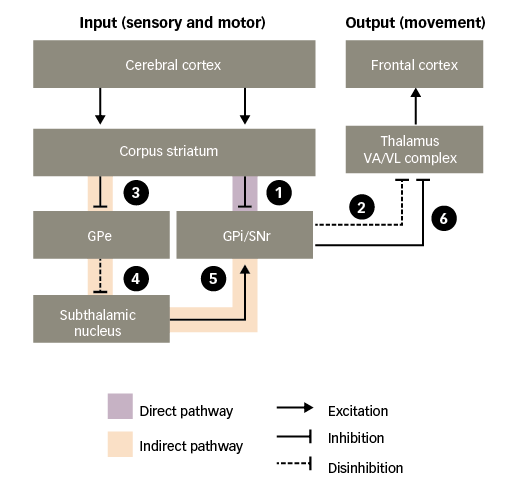
GPe = external globus pallidus; GPi = internal globus pallidus; SNr = substantia nigra pars reticulata; VA/VL = ventral anterior/ventral lateral. Adapted from Purves et al.5
In the absence of purposeful movement, the VA/VL complex of the thalamus is inhibited by the basal ganglia output nuclei (GPi and SNr), minimising its actions on the frontal cortex. Only when the inhibition is removed – termed ‘disinhibition’ – can the VA/VL complex act on the frontal cortex to stimulate motor activity. Such disinhibition of the thalamus occurs via the direct pathway:30–32
1. through the direct pathway, excitatory input to the corpus striatum from the cerebral cortex causes inhibition of the basal ganglia output nuclei; and
2. this, in turn, disinhibits the thalamic motor nuclei, leading to excitation of motor areas in the frontal cortex. Thus, the direct pathway provides a means for the basal ganglia to facilitate the selection and initiation of movement.
The indirect pathway has an opposing effect, preventing disinhibition of the thalamus:
3. through the indirect pathway, excitatory input to the corpus striatum causes inhibition of the external globus pallidus (GPe);
4. this, in turn, disinhibits the subthalamic nucleus;
5. disinhibition of the subthalamic nucleus leads to a powerful excitation of the basal ganglia output nuclei; and
6. excitation of the basal ganglia output nuclei means that the thalamic motor nuclei remain inhibited.
Thus, stimulation of the direct pathway facilitates movement, while stimulation of the indirect pathway inhibits movement. The balance of activity between the two pathways determines when the thalamus will send facilitatory signals to cortical motor areas.
Both the direct and indirect pathways are strongly influenced by dopaminergic neurons that project from the SNc to the corpus striatum, referred to as the nigro-striatal pathway (Figure 3). Dopamine acts in the striatum via two main types of receptor, called D1 and D2, which mediate facilitatory and inhibitory actions, respectively. These two types of receptors are thought to be functionally segregated, such that the D1 type is expressed in striatal neurons in the direct pathway, whereas the D2 type is expressed in striatal neurons in the indirect pathway.25,30,31,33 As a result of this organisation, dopaminergic nigro-striatal inputs have the following actions:34
1. via D1 receptors, the SNc facilitates the activation of striatal neurons in the direct pathway, increasing their responsiveness to cortico-striatal input; and
2. via D2 receptors, the SNc opposes the activation of striatal neurons in the indirect pathway, decreasing their responsiveness to cortico-striatal input.
Since the direct and indirect pathways have opposing actions on the basal ganglia output nuclei, the ultimate influence of the SNc is to lessen the inhibition exerted by the indirect pathway on the motor thalamus, therefore increasing the activity of motor areas in the frontal cortex.
As illustrated in Figure 3, glutamate mediates excitatory neurotransmission at crucial points in the basal ganglia circuitry:35
3. the corpus striatum receives an array of glutamatergic inputs from the cerebral cortex. Indeed, the corpus striatum has the highest density of glutamate receptors within the basal ganglia; and
4. the subthalamic nucleus sends glutamatergic projections to the basal ganglia output nuclei and the SNc.
The basal ganglia are a complex system with many more pathways between nuclei than have been discussed here. For example, the subthalamic nucleus also receives glutamatergic projections from the cerebral cortex (the hyperdirect pathway), and dopaminergic projections from the SNc.36 The pathways addressed here, however, are of particular interest in PD.
Figure 3: Basal ganglia functional motor circuitry – the role of neurotransmitters
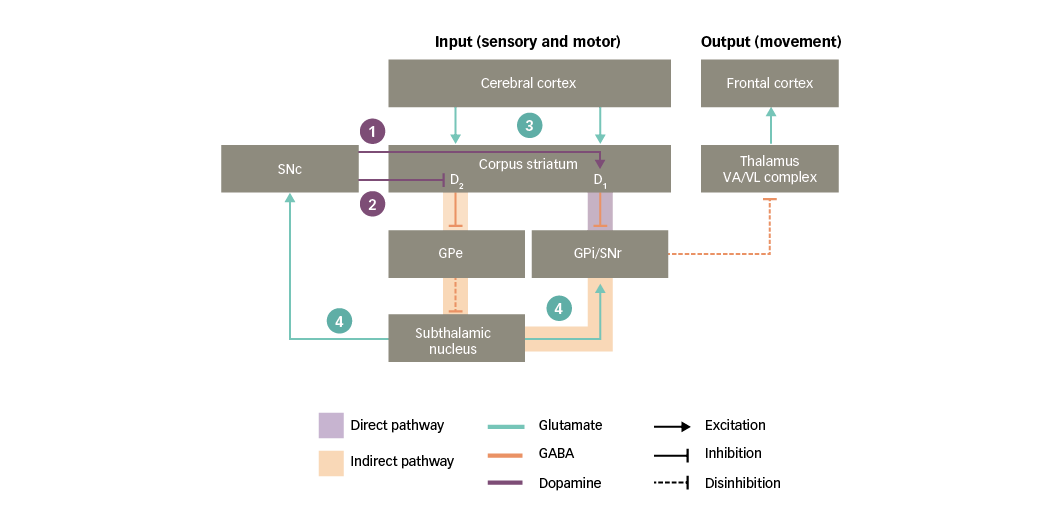
D1/D2 = dopamine type 1 or 2 receptors expressed in striatal neurons; GABA = gamma-aminobutyric acid; GPe = external globus pallidus; GPi = internal globus pallidus; SNc = substantia nigra pars compacta; SNr = substantia nigra pars reticulata; VA/VL = ventral anterior/ventral lateral. Adapted from Purves et al5 and Carrillo-Mora et al.108
Glutamate and the basal ganglia in the parkinsonian brain
Glutamate and Parkinson’s disease pathogenesis
PD is characterised by a striatal dopamine deficiency resulting from progressive loss of nigral dopaminergic neurons. As discussed in the previous section, striatal dopaminergic D1 and D2 receptors influence the direct and indirect pathways of the basal ganglia, respectively. Consequently, a chronic loss of dopamine in the striatum has the following effects on basal ganglia circuitry (Figure 4):34,37
1. dopaminergic input to the corpus striatum is reduced;
2. a deficient stimulation of D1 receptors in direct-pathway striatal neurons leads to reduced inhibition of the basal ganglia output nuclei;
3. a deficient stimulation of D2 receptors in indirect-pathway striatal neurons leads to increased inhibition of the GPe, thus resulting in disinhibition of the subthalamic nucleus;
4. further along the indirect pathway, the disinhibited subthalamic nucleus causes a glutamatergic overstimulation of the basal ganglia output nuclei (and the SNc, as discussed in the following section);
5. together, the effects on the direct and indirect pathways prevent the activation of motor thalamic nuclei; and
6. in turn, this leads to a reduced stimulation of motor areas in the frontal cortex and hence to a decreased capacity for voluntary movement.
In summary, loss of nigral dopaminergic neurons and the subsequent striatal depletion of dopamine in PD leads to glutamate overactivity in the basal ganglia where high concentrations of glutamate can damage nervous tissue.4
Figure 4: Basal ganglia functional motor circuitry in Parkinson’s disease
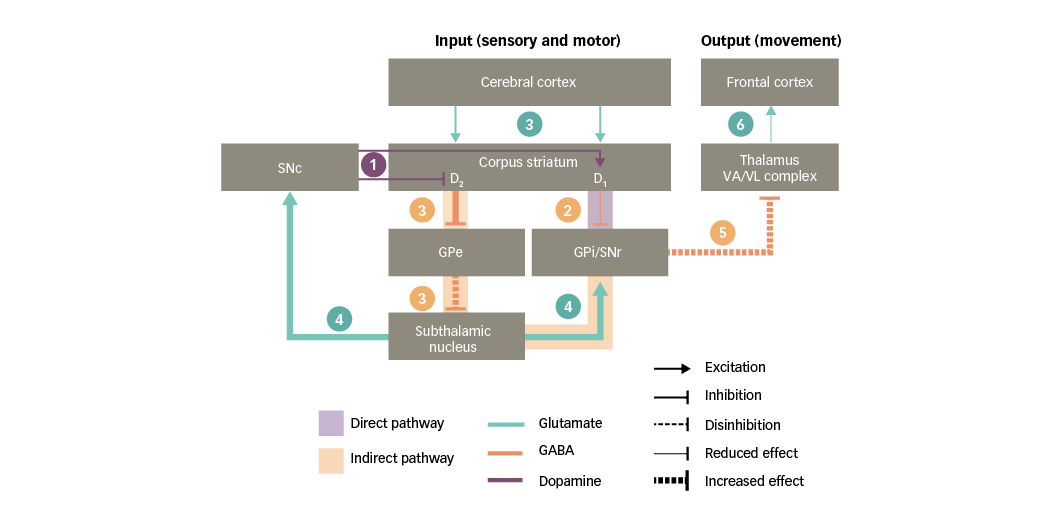
D1/D2 = dopamine type 1 or 2 receptors expressed in striatal neurons; GABA = gamma-aminobutyric acid; GPe = external globus pallidus; GPi = internal globus pallidus; SNc = substantia nigra pars compacta; SNr = substantia nigra pars reticulata; VA/VL = ventral anterior/ventral lateral. Adapted from Purves et al5 and Carrillo-Mora et al.108
Glutamate excitotoxicity and Parkinson’s disease progression
Excitotoxicity is the pathological process by which neurons are damaged or killed, mainly via excessive stimulation of glutamatergic receptors.38 The glutamate receptor subtype most strongly associated with excitotoxicity is the NMDA receptor, and the region of interest in PD is the SNc.39
The direct and indirect excitotoxicity hypotheses in PD pathogenesis stem from preclinical evidence in animal models and have to be further investigated in clinical settings.38,40
Direct excitotoxicity is the excessive stimulation of NMDA receptors due to either increased release of glutamate into the synaptic cleft, or decreased removal of glutamate from the synaptic cleft.1 In PD, glutamate overactivity in projections from the subthalamic nucleus results in glutamatergic overstimulation of the SNc.3
In early PD, this increased subthalamic nucleus activity may compensate for cell loss in the SNc by enhancing the activity of surviving dopaminergic neurons.3,41 With disease progression, however, continuous glutamatergic overstimulation of the remaining SNc neurons leads to a large influx of extracellular Ca2+ and creates metabolic changes within the neurons.4 Together, these processes have downstream intracellular consequences, notably increasing oxidative stress, disrupting mitochondrial and bioenergetic homeostasis, and activating apoptosis.4 Ultimately, therefore, glutamatergic overstimulation can result in cell death.24,39
Even in the absence of a major increase in glutamate levels, nigrostriatal dopaminergic neurons are sensitive to indirect excitotoxicity.4 Indirect excitotoxicity can arise from mitochondrial deficits; without a continuous energy supply, a cell membrane becomes depolarised, compromising the Mg2+ blockade of NMDA receptors.8,9,42 As a result, even normal levels of glutamate result in excessive stimulation of glutamatergic receptors.4,39,42 It has been hypothesised that indirect excitotoxicity may be one of multiple mechanisms contributing to PD pathogenesis, by triggering cell death in the SNc.4,39
Clinical implications of glutamate overactivity in Parkinson’s disease
The glutamate overactivity observed in PD basal ganglia clinically results both in motor symptoms and motor complications, and also non-motor symptoms.3
Motor symptoms
PD is associated with motor symptoms such as hypokinesia, bradykinesia and rigidity of the extremities and neck.37 These symptoms may arise, in part, due to glutamate overactivity in projections from the subthalamic nucleus. This leads to increased activity in the basal ganglia output nuclei that inhibit the thalamus, resulting in reduced facilitatory input to motor areas in the frontal cortex (Figure 5).38 As a result, movements are difficult to initiate and, once initiated, they are slow and may be difficult to stop.
Figure 5: Basal ganglia functional motor circuitry in Parkinson’s disease, and potential links to motor symptoms of Parkinson’s disease
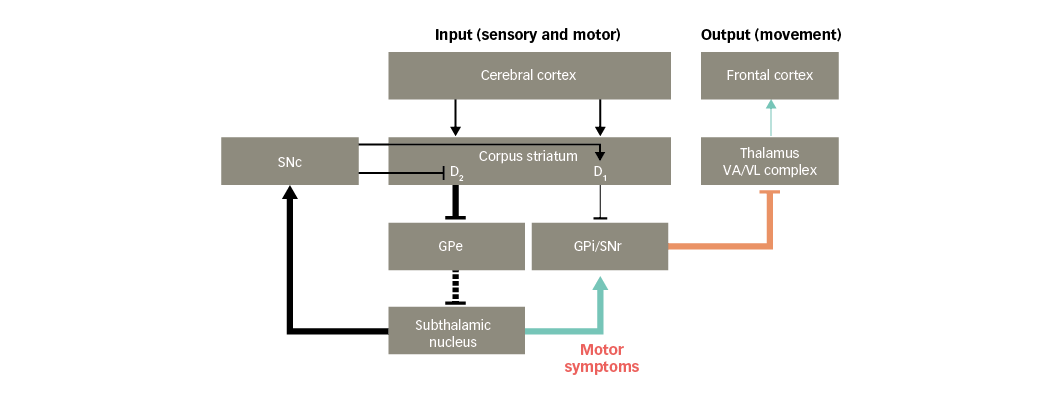
D1/D2 = dopamine type 1 or 2 receptors expressed in striatal neurons; GPe = external globus pallidus; GPi = internal globus pallidus; SNc = substantia nigra pars compacta; SNr = substantia nigra pars reticulata; VA/VL = ventral anterior/ventral lateral. Adapted from Finlay et al.110
Motor complications and levodopa-induced dyskinesia
Levodopa, a dopamine precursor, is used as a pharmacotherapy in PD to enhance dopaminergic neurotransmission. Over time, this benefit may wane and patients start to notice loss of benefit and re-emergence of PD symptoms with each dose of levodopa (wearing-off phenomenon) and fluctuate between mobility and immobility (on and off state, respectively). With chronic use of levodopa, furthermore, patients may experience troublesome involuntary movements – the so called levodopa-induced dyskinesia (LID) – that severely impact upon quality of life.43,44
Chronic levodopa treatment is thought to lead to overactivity of the direct pathway and underactivity of the indirect pathway, ultimately resulting in cortical excitation and dyskinesia.43 Overactivity of glutamatergic corticostriatal projections is a critical factor in the overactivity of the direct pathway, contributing to both the development and expression of LID.43,45However, the pathophysiology of LID is complex, and a variety of neurotransmitters and receptors are involved in addition to glutamate.46,47
Non-motor symptoms
In addition to the motor symptoms already described, PD is associated with a range of non-motor symptoms, including pain, cognitive impairment/dementia, and mood disorders.48 Such non-motor symptoms are common, can occur at any stage of the disease, and are key determinants of quality of life.
Glutamate, as well as being the major excitatory neurotransmitter in the CNS and being key to learning and memory, also plays an important role in peripherally mediated pain signalling to the CNS49 and in mood control.50 This section considers the possible contribution of glutamate overactivity to the occurrence of specific non-motor symptoms in PD.
Pain
Pain has a prevalence of around 60% among patients with PD.51,52 Pain in PD is most often of nociceptive origin, but may also be ascribed to neuropathic (radicular or central) or miscellaneous sources. While there are various ways to classify pain in PD, the most widely accepted is the Ford classification, which distinguishes five different types of PD pain: musculoskeletal (felt as an ache around the joints, arms or legs), dystonia-related, radicular or neuropathic, primary/central, and akathisia.51 Indeed, the recently validated King’s Parkinson’s Disease Pain Scale is based on seven domains including musculoskeletal pain, chronic body pain (central or visceral), fluctuation-related pain, nocturnal pain, oro-facial pain, pain with discolouration/oedema/swelling, and radicular pain.53 Patients with PD who report pain symptoms are also more likely to report depression and a decreased quality of life.51 Many patients with PD also report poor management of pain, with the use of analgesics being lower than expected (around half of patients).51 More recent findings showed that patients with PD both with and without pain may have a low heat pain threshold (regardless of being in an ‘on’ or ‘off’ state), and abnormal pain-evoked responses suggest that patients with PD may be predisposed to developing pain.54,55
Large and case-control clinical studies found that a consistent proportion of patients experienced pain when they were drug-free.56–58 This supports a link between pain and pathophysiological PD mechanisms. Because basal ganglia are involved not only in motor functions but also in the processing of nociceptive and non-nociceptive inputs, it is conceivable that nigrostriatal damage leading to a dysfunction of the control exerted by basal ganglia on cerebral areas devoted to processing nociceptive inputs might at least partly account for the increased risk of pain in PD.59 Nigral and extra-nigral pathology, involving cortical areas, brainstem nuclei and spinal cord, may contribute to abnormal central nociceptive processing in patients with PD who are experiencing pain.53 Recently, the contribution of the dorsal striatum in pain inhibition has been demonstrated.60,61 The dorsal striatum is in effect connected to the descending pain modulatory system and in particular to the rostral ventromedial medulla through the medullary dorsal reticular nucleus.62The neurobiology of pain in PD is complex and appears to involve serotonergic, noradrenergic, glutamatergic and GABAergic neurotransmission, in addition to the dopaminergic systems.63
Glutamate neurotransmission plays an important role in both normal and pathophysiological nociception.64 Peripheral sensory information and pain signals are transmitted to the spinal cord via primary afferent neurons, the majority of which are glutamatergic. Upon noxious stimulation, glutamate is released from central terminals in the spinal cord, where it activates AMPA receptors on secondary neurons. Prolonged activation of nociceptors evokes continuous release of glutamate, and subsequently causes long-lasting membrane depolarisation. This exaggerated signalling relieves the voltage-dependent Mg2+ block on NMDA receptors, and consequently allows their activation by glutamate. Additionally, postsynaptic mGluRs, (specifically mGluR1 and mGluR5),65 and some presynaptic kainate receptors localised on central terminals of primary afferents, also play a role in nociceptive transmission. Persistent peripheral stimuli or damaged primary afferents trigger a cascade of events that increase the efficacy of glutamatergic neurotransmission in the spinal cord, a phenomenon known as central sensitisation. A key molecular mechanism of central sensitisation is differential sensitisation of AMPA receptors and NMDA receptors and enhanced ion channel activity. Central sensitisation is thought to underlie the progression of chronic pain, including the effects of allodynia and hyperalgesia.66Antagonists of ionotropic glutamate receptors, including NMDA, AMPA and kainate receptor antagonists, decrease nociceptive transmission,67but have a narrow therapeutic window due to their side effects. However, the development of selective mGluR ligands has provided important tools for further investigation of the role of glutamate in the modulation and control of chronic pain processing.68
Elevated glutamatergic neurotransmission in the CNS is observed during neuropathic pain, which is accompanied by lower effectiveness of opioid anti-nociceptive drugs.69 Modulation of glutamatergic system activity could be beneficial for the potentiation of an analgesic effect of opioid drugs in neuropathic pain, and possibly for other drugs used in the treatment of neuropathic pain, such as antidepressants. Recently, it was shown that opioid-induced hyperalgesia in neuropathic pain could be inhibited by upregulation of spinal glutamate transporter-1 (GLT-1) expression.70 Moreover, the effect of other drugs used in neuropathic pain therapy could be potentiated by the use of mGluR ligands and/or modulators of EAATs. Recently, it has been demonstrated that the dorsal striatal mGluR7, by modulating glutamate and GABA release, may play an important role in the supraspinal pathways involved in the control of pain.71 The stimulation of mGluR7 in the dorsal striatum decreased glutamate levels in both control and neuropathic rats, while it inversely modulated the pain and neural activity of the rostral ventromedial medulla and dorsal reticular nucleus. The opposite effects of mGluR7 stimulation in normal and neuropathic pain conditions was thus likely associated with the involvement of different pathways from the dorsal striatum to dorsal reticular nucleus and rostral ventromedial medulla. In control rats, the mGluR7 stimulation involves the indirect pathway of the basal ganglia, whereas in the neuropathic conditions, the direct pathway of the basal ganglia. Therefore, mGluR7 in the dorsal striatum in neuropathic conditions functions as an ‘emergency brake’ and reduces pain responses (and neural hyperactivation in the rostral ventromedial medulla and dorsal reticular nucleus) by the direct pathway, the activity of which is likely enhanced in pathological pain conditions.
Cognitive impairment and dementia
Mild cognitive impairment (MCI) and dementia are common non-motor symptoms in PD.72,73 MCI is common in PD, even in early stages of the disease; about 20% of patients with PD meet the criteria for MCI at the time of diagnosis, and MCI is strongly associated with a progression to dementia.74 Patients with PD have an approximately sixfold increased risk for developing dementia compared with neurologically healthy elderly individuals.75 The prevalence of dementia in PD is around 25–30%,76 but up to 80% of patients with PD will progress to dementia after 15–20 years.77,78 MCI presents as a difficulty in recalling facts, finding words, and concentrating, while dementia presents as severe problems in thinking and memory that interfere with the ability to undertake daily activities.72,73
Given the heterogeneous neuropathological and neuropsychological nature of cognitive deficits, it has been hypothesised that there are two independent, partially overlapping syndromes in PD:
- a frontal-striatal network dysfunction present at the early stage of the disease, which is dopamine-modulated, leading to deficits in working memory, attention, planning and response inhibition, and responds to dopaminergic therapy; and
- an additional, more posterior cortical degeneration, which is associated with cholinergic loss and wherever present would lead to dementia, responding better to cholinergic therapy.79,80
Specific brain circuits are believed to be involved in the development of MCI in patients with PD.72 Impairments in executive functions may be attributed to failure in the frontal-striatal basal ganglia circuits, known to be affected in PD.72 Visuospatial and memory deficits, in contrast, are thought to arise from a decline in posterior cortical functioning.72Decreased activity in neuronal circuits connecting the thalamus and the cortex results in reduced glutamate input to specific frontal cortical areas.34
Glutamate is involved in most, if not all, aspects of cognition and higher mental functions.2 Decreases in cortical glutamate, together with decreased striatal dopamine availability,81 may underlie the mechanisms of decline in frontal-cortex-based cognitive functions, such as in PD. Glutamate has an extensive role in memory encoding and maintenance.82 In some specific areas of the brain (such as hippocampal regions), the activation of NMDA receptors is crucial for long-term potentiation, a cellular correlate of learning and memory formation.82 Problems with learning and memory can arise due to reduced NMDA receptor system function as the brain ages, resulting in an excess of extracellular glutamate that can lead to severe cognitive impairments and psychosis.83
Glutamate excitotoxicity at NMDA receptors may be involved in dementia observed in patients with PD. The potential role of dysregulated glutamate in cognitive impairment is supported by studies of NMDA receptor antagonists (such as memantine), which reduce glutamatergic signalling.84
Mood disorders
Mood disorders, particularly depression and anxiety, are common in PD and can pre-date motor symptoms. Mood disorders have a significant negative impact on a patient’s prognosis and quality of life.85 Around 35% of patients with PD have clinically significant depressive symptoms and approximately 17% have a concurrent diagnosis of major depressive disorder, although incidences vary between studies.86 The prevalence of anxiety disorders in PD is about 31%.87
The pre-motor symptoms of depression and anxiety are thought to relate to brainstem monoamine pathology affecting the locus coeruleus and dorsal raphe nucleus at an early stage, before the occurrence of dopamine cell loss in the SNc.88,89 Depression and anxiety, as observed in later PD, probably reflect multiple neurotransmitter changes (notably monoamines such dopamine, noradrenaline, and serotonin) and quite recently, preclinical and clinical studies have also provided evidence of the involvement of glutamatergic and GABAergic transmission.64,90,91
Perturbation of the glutamatergic system is considered to be at least partially involved in the synaptic abnormalities found in mood disorders,92 particularly a deficit of glutamate clearance at the synaptic space.93Proof-of-concept regarding the involvement of the glutamatergic system in mood disorders was based on clinical data with ketamine, an NMDA antagonist found to exert a rapid antidepressant effect in patients with depression,94thus moving from the monoamine hypothesis to the neuroplasticity hypothesis of mood disorders, integrating glutamatergic signalling, gene expression, neurotrophic mechanisms, neurogenesis, and synaptic plasticity.95,96
Several findings from postmortem studies are in accordance with the evidence of glutamate abnormalities in mood disorders.97 Moreover, magnetic resonance spectroscopy studies in patients with depression found increased levels of glutamate and/or glutamine – the metabolite and precursor of glutamate – in the basal ganglia.98,99 In particular, the increase in glutamine in putamen is of interest in view of the postulated role of the basal ganglia in the neuropsychology of depression, and is consistent with elevated activity in the descending cortical glutamatergic innervation to the putamen. Striatum, a reward-related brain area, is enriched with glutamatergic afferents and glutamate receptors and is involved in depression-related behaviour such as anhedonia. In preclinical depression models,100 striatal mGlu5 receptor expression was increased, leading to hyperactivity of mGlu5 receptor signalling, which thereby contributes to depressive-like symptoms that are prevented by mGluR5 antagonists.101–103
It is worth mentioning that episodes of hypomania/mania have been reported in patients with PD, due to hyperstimulation of the dopaminergic system.104,105 There is also evidence that the limbic target areas of the ventral frontal cortex show increased neuronal activity when patients are in the manic state.106 Since these projections are glutamatergic, mania-related overactivity may be suggestive of excessive activation of glutamatergic cortico-limbic pathways. Indeed, mood stabilisers, such as lithium and anticonvulsant voltage-gated Na+/Ca2+ channel blockers, are effective in treating symptoms of both depression and mania, and appear to attenuate glutamatergic functions by reducing glutamate release, postsynaptic excitability and intracellular signalling downstream from glutamate receptors.107
Glutamate as a potential target for Parkinson’s disease pharmacotherapy
Glutamate is implicated in various aspects of PD – from motor and non-motor symptoms, to treatment-related complications and even to its pathogenesis.1,3,64 Consequently, the glutamatergic system is a logical target for pharmacotherapies in PD.108 This section summarises the evidence for whether the action of drugs on the glutamatergic system has translated into a clinical effect.
Traditional pharmacotherapies used in the early stages of PD aim to increase nigrostriatal dopaminergic neurotransmission, thereby preventing glutamatergic overstimulation of the basal ganglia output nuclei by the subthalamic nucleus. Such therapies include levodopa, dopamine agonists, and drugs that inhibit the breakdown of dopamine by monoamine oxidase B (MAO-B) or the breakdown of levodopa by catechol-O-methyltransferase (COMT).109
However, as a result of striatal dopaminergic denervation, the glutamatergic projections from the subthalamic nucleus to the basal ganglia output nuclei become overactive with reduced regulation of glutamate receptors. The resultant excessive excitation by glutamate through the basal ganglia circuitry can be toxic to any remaining dopaminergic neurons, leading to PD motor symptom exacerbation and impaired output to cortical regions (non-motor symptoms).34,37 In addition, dopamine-enhancing treatment with levodopa induces severe motor complications such as motor fluctuations (wearing off) and dyskinesia, where abnormal glutamate transmission plays an important role.43–47
A number of pharmacotherapies are available or are being tested in clinical trials that directly target the glutamatergic system in PD.39,108,110–112 Such drugs may act on different sites or processes within the tripartite glutamatergic synapse, including glutamate receptors, glutamate release and glutamate uptake. These targets and agents are discussed in more detail below.
Modulation of glutamate receptors
Effect on motor symptoms and levodopa-induced dyskinesia
NMDA receptor antagonists block NMDA receptor activity, thereby reducing NMDA receptor overstimulation by glutamate, and preventing excitotoxicity.8,38,110,111 Amantadine is a nonselective NMDA antagonist currently in clinical use to treat LID; however, it is associated with side effects including confusion and visual hallucinations.111 Longer-acting formulations such as amantadine extended release capsules (former ADS-5102), may be able to improve the side-effect profile by reducing night-time drug levels; phase III studies have shown consistent and long-lasting reductions in LIDs.113,114 Moreover, in small phase II/III studies, NMDA receptor antagonists such as dextromethorphan/quinidine (a combination drug) and memantine have shown some clinical benefits on LID (decreased severity/peak dyskinesia and reduced time with dyskinesia, respectively).112,115
Antagonists that block only the NR2B subtype of NMDA receptor are also under investigation; preliminary clinical results are suggestive of a benefit on dyskinesia.116
In contrast, AMPA receptor antagonists such as perampanel have not shown a consistent clinical benefit in PD and are no longer being pursued as a therapy to treat motor symptoms.117,118
mGluRs have an indirect modulating effect on glutamatergic pathways. The mGluR5 subtype of group I receptor is of particular interest in the treatment of PD since it is functionally connected to both D2 and NMDA receptors, and is highly expressed in the basal ganglia.119 mGluR5 appears to amplify the effects of NMDA receptor stimulation, while NMDA receptors may amplify the effects of mGluR5 activity – a reciprocal positive feedback interaction that could contribute to NMDA-receptor induced excitotoxicity.120,121 Although preliminary clinical results are suggestive of a benefit of mGluR5 negative allosteric modulators on dyskinesia, there are currently no known plans for future studies (e.g., of mavoglurant or dipraglurant) in PD.111
At the striato-pallidal and subthalamopallidal synapses, mGluR4 activation reduces GABA and glutamate release in both pallidal segments and is predicted to modulate the overall activity of the overactive indirect pathway in PD.122 At the corticostriatal synapses, mGluR4 decreases excitatory transmission from the cortex. Foliglurax, a mGluR4-positive allosteric modulator, alleviates motor symptoms and LID in PD primate models, demonstrated good safety and tolerability in phase I studies and is now being assessed in patients with PD in a phase II study.123
Effect on non-motor symptoms
Preclinical studies have shown that NMDA antagonists were effective in models of pain induced by inflammation, tissue and nerve injury and visceral nociception. Further, clinical studies in patients with neuropathic pain have found that the NMDA receptor antagonist ketamine can be used to control neuropathic pain.124 In short-term clinical trials in mood disorders, ketamine and the NR2B receptor antagonist, traxoprodil (former CP-101,606), have also shown benefits in patients with depression.125,126 However, NMDA antagonists have not yet been tested in patients with PD and depression or pain.
With regard to cognition, memantine, an NMDA receptor antagonist, has been shown to reduce the rate of deterioration among patients with PD dementia.127 However, there is some disagreement between studies.128,129 In a very recent meta-analysis, memantine was revealed to have some beneficial effects on attention, processing speed and executive functions in patients with PD and cognitive impairment, dementia and dementia with Lewy bodies.130 However, considering the limitations of the study, the authors concluded that the results did not sufficiently support the use of memantine as treatment for PD cognitive impairment and PD dementia.
An observational study in patients with PD showed that amantadine, another NMDA receptor antagonist, may delay the onset of PD dementia and attenuate its severity.131
Inhibition of glutamate release
Effect on motor symptoms and levodopa-induced dyskinesia
As described above, in response to an action potential, voltage-gated Na+ channels on the presynaptic neuron open to allow the passage of Na+ into the neuron. The resulting cell membrane depolarisation causes an influx of Ca2+ through voltage-gated Ca2+ channels, which stimulates vesicles to fuse with the membrane, thereby releasing glutamate into the synaptic cleft.
Both Na+ and Ca2+ channels are targets for pharmacotherapy. Agents that block voltage-gated Na+channels and agents that block voltage-gated Ca2+ channels may indirectly or directly inhibit the presynaptic release of glutamate, respectively.132,133 Safinamide, a drug that reduces the abnormal glutamate release through use-dependent Na+ channel blockade134 and inhibits MAO-B, has shown in add-on to levodopa a clinical benefit by reducing ‘off’ time with no worsening of troublesome dyskinesia likely due to its anti-glutamatergic activity.104,135–137 Zonisamide, a Na+ channel blocker and weak MAO-B inhibitor, when added to levodopa therapy was found to be beneficial in treating motor symptoms in Japanese patients with PD.138
Effect on non-motor symptoms
In open-label studies and inpost hoc analyses, agents that block voltage-gated Na+ and Ca2+ channels, thereby inhibiting glutamate release, have shown promise for the treatment of peripheral and central neuropathies and mood disorders, cancer pain, and HIV-associated pain.125,139 Safinamide has been shown to improve PD-related chronic pain and mood fluctuations.140–142
Modulation of glutamate uptake
Dysfunctions in glutamate uptake due to downregulation of the high-affinity astrocytic glutamate transporters (EAAT2/GLT-1 and EAAT1) results in increased availability of glutamate in the synaptic cleft.5Preclinical studies have provided some insight into how elevated glutamate levels in specific brain regions may account for motor symptoms, depression- and anxiety-like behaviour, and pain in animal models.143,144 These symptoms have been prevented and/or reversed by the GLT-1 inducer, ceftriaxone.145–148
Concluding remarks
Glutamatergic neurotransmission in the basal ganglia is essential for the normal control of movement, as well as for pain, cognition and mood. When the striatum receives excitatory glutamatergic input from the cerebral cortex, two pathways are activated whose co-ordinated action enables normal motor activity.
In PD, however, the degeneration of nigrostriatal dopaminergic neurons causes chain reactions that ultimately result in a glutamatergic overstimulation of the basal ganglia output nuclei and the SNc. Overstimulation of the basal ganglia output nuclei leads to parkinsonian motor symptoms, in particular, hypokinesia, bradykinesia and rigidity. Glutamate overactivity may contribute to the emergence of LID, as well as non-motor symptoms such as pain, cognitive impairment and depression. In addition, overstimulation of the SNc can lead to excitotoxicity, which might play a role in the pathogenesis and progression of PD.
Research into glutamate signalling in the basal ganglia is revealing a complex and interconnected network involving cross-talk between glutamate and dopamine transmission. The knowledge of these interactions will enable to develop compounds that can fine tune dopaminergic and non-dopaminergic transmission, leading to better treatments for PD. Thus, glutamatergic neurotransmission is a potential focus for pharmacotherapy in PD. A number of drugs have glutamate-related targets and these therapies have the potential to improve both motor and non-motor symptoms of PD. ⬛