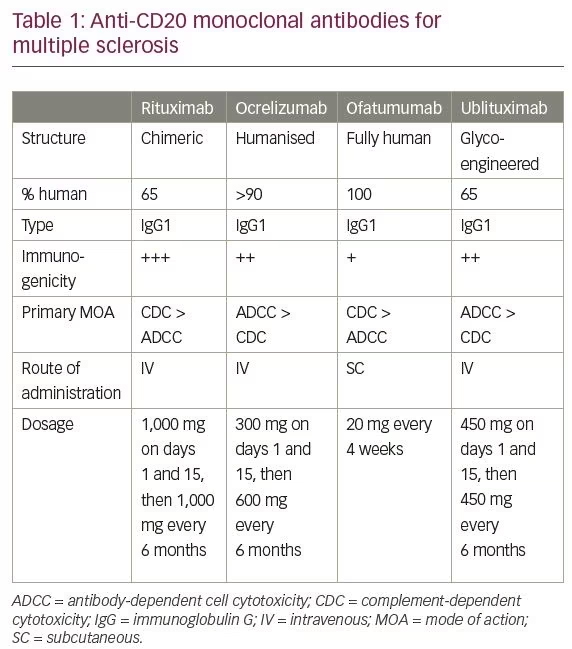
Multiple sclerosis (MS) has traditionally been considered a primarily T-cell-mediated autoimmune disease of the central nervous system (CNS); this is based on data from animal models,1 the presence of activated T lymphocytes in MS plaques that outnumber B cells and T-cell subset alterations in blood,2 the association of MS with certain human leukocyte antigen (HLA) and a number of other alleles critical for T-cell activation that have been identified in genome-wide association studies.3,4 The role of B cells in the pathogenesis of MS has been ignored for many years, despite the discovery of oligoclonal immunoglobulins in the cerebrospinal fluid (CSF) of patients with MS 60 years ago,5 the presence of clonally-expanded B cells and plasma cells in MS lesions,6 and the observed immunoglobulin deposition and complement activation in active demyelinating lesions in some patients with MS7 that is capable of inducing demyelination.8 Recent advances in flow cytometry and DNA sequencing methods have revealed the fundamental contribution of B cells, plasma cells and their products to immune responses, and to the pathogenesis of MS and other immune-mediated disorders.9 However, the most compelling evidence for the central role of B cells in MS came from clinical trials with B cell-depleting-agents that demonstrated a dramatic therapeutic effect in patients with relapsing forms of MS, particularly anti-CD20 monoclonal antibodies (mAbs).10 CD20 is a transmembrane ion-channel non-glycosylated phosphoprotein expressed on cells of the B-cell lineage from pre-B cells to memory B cells and early plasmablasts, but not on stem cells, pro-B cells, late plasmablasts, or antibody-producing plasma cells. Anti-CD20 mAbs deplete target cells by induction of four main mechanisms: antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP) and induction of direct cell death (erroneously often called apoptosis).11 Four such antibodies which act mainly through CDC and ADCC mechanisms have been studied in MS: the first-generation chimeric mAb rituximab; the second-generation humanised mAb, ocrelizumab, currently registered for both relapsing and primary progressive MS (PPMS); the third-generation fully-human mAb, ofatumumab; and the third-generation glycoengineered chimeric mAb, ublituximab (Table 1).
Treatment with rituximab demonstrated a profound anti-inflammatory effect and >80% reduction in relapse rate in a phase II clinical trial in patients with relapsing–remitting MS (RRMS).10 In patients with PPMS, rituximab was able to reduce disability progression only in a subgroup of patients younger than 51 years who had enhancing lesions on baseline magnetic resonance imaging (MRI).12 The development of rituximab was discontinued for a variety of reasons and attention has been shifted to less immunogenic and potentially more potent anti-CD20 mAbs; however, rituximab is still being used off-label for MS.13 Ocrelizumab is approved for both relapsing and progressive forms of MS, after showing high efficacy and superiority to subcutaneous (SC) interferon (IFN)-β-1a in relapsing MS,14 and reduced rate of disability progression in PPMS.15 Recently, positive results from a phase II clinical trial with ublituximab16 and two phase III trials with ofatumumab in relapsing MS have been announced.17 This review will focus on the development of ofatumumab for the treatment of patients with MS.
Role of B cells in multiple sclerosis
According to the traditional view, T cells are playing a key role in the immune pathogenesis of MS, where an abnormal balance between CNS-reactive effector CD4+ Th1/Th17 cells and regulatory T cells (Treg) underlies autoimmunity directed at the CNS.18 In this scenario, different T-cell subsets can also shape immune responses of myeloid cells that can be polarised towards either pro-inflammatory macrophages secreting interleukin (IL)-12, IL-23, IL-6 and IL-1β or anti-inflammatory macrophages secreting IL-10, while in turn their own responses can be shaped by differentiated macrophages. B cells were considered to be a relatively passive population, awaiting the help of T cells to differentiate into plasmablasts and plasma cells that contribute to MS pathophysiology by producing CNS-autoreactive antibodies.19 An emerging view, however, looks at B cells as having a more central and broader role in MS, which is mainly antibody-independent. Recent research has shown that B cells can have several phenotypes according to their cytokine profile, and manifest as either pro-inflammatory effector B cells (including memory B cells that are increased in MS) secreting tumour necrosis factor (TNF)-α, lymphotoxin (LT)-β, IFN-γ, IL-6, IL-15 and granulocyte macrophage colony stimulating factor (GM-CSF); or anti-inflammatory regulatory B cells (Breg), secreting IL-10, transforming growth factor (TGF)-β and IL-35, that either activate or downregulate responses of both T cells and myeloid cells. According to this view, CNS autoimmunity is shaped by complex bidirectional interactions among functionally distinct populations of T cells, B cells and myeloid cells, some of which may be over-active or hypo-functional in MS.19
Abnormally elevated levels of immunoglobulins and the presence of oligoclonal bands in the CSF of most patients with MS indicate the intrathecal production of immunoglobulins by a restricted number of expanded clones of B cells and plasma cells. These clones persist within the CNS in individual patients over time and can be shared among different CNS and peripheral compartments, suggesting bidirectional trafficking of distinct B-cell clones between the CNS and the periphery.19 B cells can get access into the CNS across the blood–brain barrier via parenchymal vessels into the perivascular space and via post-capillary venules into the subarachnoid and Virchow–Robin spaces. They can also cross the blood–CSF barrier via the choroid plexus into the CSF, and via the blood–leptomeningeal interphase.20 The recently-discovered functional lymphatic vessels lining the dural sinuses can transport CSF and lymphocytes, including antigen-carrying B cells, mainly into the deep cervical lymph nodes where much of the clonal expansion of these B cells occurs.21 Thus, B cells can dynamically traffic into and out of the CNS in MS; process and present CNS antigens in the deep cervical lymph nodes; make their way back into the CNS via the thoracic duct, systemic circulation and the various brain barriers; infiltrate the brain parenchyma; populate ectopic lymphoid follicles; and trigger another bout of CNS-targeted inflammation.20
B cells can contribute to the pathogenesis of MS and to CNS tissue damage by several mechanisms, including antibody production, antigen presentation, modulation of T-cell response including the activation of autoproliferating CD4+ T cells, cytokine production, and formation of ectopic germinal centres.
Antibody production
The antibodies that make up the oligoclonal bands are produced by plasma cells generated from a restricted number of B-cell clones that persist within the CNS of the same individual, but differ among patients.22 They primarily recognise ubiquitous intracellular proteins, but no specific antigens that are shared across patients with MS, suggesting a humoral response to debris from dead cells rather than a primary pathogenic response.23 In addition, the lack of association between anti-myelin or anti-KIR4.1 antibodies found in patients with MS and disease progression,24,25 and the rapid decrease in clinical and MRI disease activity after B-cell depletion10,14 that is unlikely to result from removal of any pathogenic antibodies that have relatively long half-life, also support no major pathogenic role for antibodies produced by B cells and plasma cells in MS. On the other hand, demyelinating MS lesions contain immunoglobulins and activated complement, which may suggest antibody-mediated damage, at least in some patients,7 and anti-myelin oligodendrocyte glycoprotein (MOG) antibodies have been shown to contribute to demyelination in the experimental allergic encephalomyelitis (EAE) model26 and in anti-MOG disease in humans.
Antigen presentation
B cells are highly effective antigen-presenting cells (APCs). They express high levels of major histocompatibility complex (MHC) molecules on their surface, which are essential for antigen presentation to T cells. B cells also express membrane-bound antigen-specific immunoglobulins, which correspond to the soluble immunoglobulins they secrete after developing into plasmablasts or plasma cells. These B-cell receptors (BCRs) recognise and bind three-dimensional conformational epitopes, compared with other APCs such as dendritic cells and macrophages that present short linear epitopes.27 Internalisation of the BCR-antigen complexes allows for the stimulation of MHC-II synthesis, which are then loaded with linear antigenic peptides after degradation and processing of the BCR-antigen complexes. Antigen presentation by resting B cells promotes T-cell tolerance, while activation of B cells by antigen and T cells promotes immune responses. The high efficacy of B cells in antigen presentation, particularly when they recognise the same antigen as T cells (cognate recognition), makes them the main source of APCs when antigen levels are low.27 In EAE, antigen presentation by B cells is critical for disease induction, independent of their antibody specificity.28
In MS, the number of circulating B cells expressing the co-stimulatory molecule CD80 that interacts with CD28 on T cells for efficient antigen presentation is increased significantly during MS exacerbations, but is normal in stable MS.29 On the other hand, the association of the co-inhibitory molecule programmed death ligand 1 (PD-L1) on B cells with programmed cell death protein 1 (PD-1) on T cells downregulates T-cell responses and protects against EAE, and expression of the co-inhibitory glucocorticoid-induced TNF ligand (GITRL) on B cells induces Treg cells and suppresses autoimmunity through its receptor, GITR.30
Activation of autoproliferating autoreactive CD4+ T cells
Autoproliferation is the activation and growth of myelin-specific T cells by APCs in the absence of exogenous antigen, which was found to be increased in patients with MS.31 This autoproliferation was found to be driven by memory B cells in a HLA-DR-dependent manner in patients with MS carrying the HLA-DR15 haplotype and to be reduced by B-cell depletion with anti-CD20.32 The autoproliferating CD4+ T cells are enriched for brain-homing, probably pathogenic T cells, and recognise a target autoantigen – RASGRP2 – that is expressed in both the brain and B cells.32 These data may also explain, in part, the high efficacy of anti-CD20 therapy in a T-cell-mediated disease such as MS.
Cytokine production
B cells can produce both pro- and anti-inflammatory cytokines, and can modulate local immune responses according to their cytokine profile. In MS, B cells are skewed toward a pro-inflammatory cytokine profile and produce abnormally high amounts of pro-inflammatory cytokines (e.g., IFN-γ, TNF-α, LT-α, IL-6 and GM-CSF), which may activate T cells and myeloid cells and contribute to the disease process.33 B-cell autoimmunity may also be enhanced by B-cell activating factor (BAFF) secreted in high amounts in MS lesions from astrocytes stimulated by TNF-α produced by these B cells. The close interactions between T cells, B cells and myeloid cells, and the contribution of cytokines secreted from pro-inflammatory B cells to MS pathogenesis, independent of their antibody-production function, are also highlighted by the abrogation of inflammatory responses of all three cell types with B-cell depletion using anti-CD20 mAbs.19 In addition, Breg secreting anti-inflammatory cytokines (e.g., IL-10, TGF-β and IL-35), that can downregulate immune responses and limit neuroinflammation, were found to be defective in MS.34
Formation of ectopic germinal centres
In diseases characterised by chronic inflammation such as MS, B cells that have been attracted to the brain under stimuli such as the chemokine CXCL13 with the appropriate help from T cells can proliferate, aggregate and generate meningeal inflammation and eventually ectopic immunocompetent germinal centre-like structures, also called tertiary lymphoid structures, in the leptomeningeal space adjacent to the cortex of the brain that are associated with more severe cortical pathology and more aggressive disease course.35 These B-cell-rich ectopic lymphoid structures have been described mainly in SPMS,35 but can also be found in RRMS36 and PPMS,37 and can potentially serve as reservoir of memory B cells, autoreactive plasmablasts and plasma cells that perpetuate autoimmune disease. It has been suggested that soluble factors released from B-cell follicles adjacent to subpial cortical lesions can be cytotoxic to both oligodendrocytes38 and neurons39 and contribute to cortical injury in MS.
The role of Epstein–Barr virus (EBV) and EBV-infected B cells in the pathogenesis of MS is controversial. The risk for developing MS is markedly increased in patients who have had late infection with EBV and infectious mononucleosis, and in patients with MS, sero-conversion to EBV nuclear antigen-positive antibody status occurs always prior to the first appearance of MS symptoms. EBV is known to infect and immortalise B cells. Potential mechanisms underlying the association between EBV infection and MS may include the activation and expansion of autoreactive T and B cells during infectious mononucleosis, a syndrome characterised by strong immune activation, or the ability of EBV-immortalised B cells to produce autoantibodies and present antigens to pathogenic T cells. In addition, EBV could assist in the survival of autoreactive B cells in MS, thus promoting neuroinflammation.40 However, there is no experimental evidence supporting these hypotheses.
Ofatumumab
Ofatumumab is a fully-human, high-affinity immunoglobulin G (IgG)1κ mAb with a very low immunogenic risk profile that binds strongly to, and dissociates more slowly from, epitopes on both the small and large extracellular loops of the CD20 molecule on B cells that are completely distinct from the overlapping epitopes of rituximab and ocrelizumab and are closer to the cell membrane. This proximity is thought to increase the activity of ofatumumab through a CDC mechanism, which might be dependent on the distance between the plasma membrane and the antibody.41 Moreover, ofatumumab may have a greater potential for effector activity, particularly on cells expressing low levels of CD20.41 Ofatumumab was initially developed by GlaxoSmithKline (GSK; Brentford, UK) and Genmab AS (Copenhagen, Denmark) for chronic lymphocytic leukaemia (CLL) and additional indications in oncology, and for the autoimmune diseases rheumatoid arthritis (RA), MS, pemphigus vulgaris and chronic obstructive lung disease (COPD). Intravenous ofatumumab (Arzerra®, Novartis, Basel, Switzerland) is approved for the treatment of patients with refractory CLL. The development in RA and COPD was discontinued by GSK. In 2015, Novartis acquired the rights for the development and commercialisation of ofatumumab in all indications and subsequently decided to discontinue the development in pemphigus vulgaris and focus on the development of ofatumumab in MS.
Pharmacokinetics and pharmacodynamics
SC ofatumumab developed for simplified administration has been demonstrated to be well tolerated in a small study where the pharmacokinetics (PK) and pharmacodynamics (PD) of SC ofatumumab have also been investigated in 35 patients with RA.42 The PK of SC ofatumumab was shown to be typical of a mAb with low clearance, a small volume of distribution, and a relatively long half-life. A single SC low dose of ofatumumab at 0.3 mg or 3.0 mg resulted in plasma ofatumumab concentrations near or below the lower limit of quantification. After a single SC dose of 30 mg, 60 mg or 100 mg, ofatumumab was slowly absorbed, with maximum plasma concentration values ranging from 827 to 3,405 ng/mL, median Tmax values ranging from 4.02 to 4.49 days and mean elimination half-life values ranging from 5.20 to 6.83 days. PK studies indicated an increasing level of CD19+ B-cell depletion from the 0.3 mg to the 30.0 mg group, with profound (≥95%) and sustained CD19+ B-cell depletion achieved in all three higher-dose cohorts. Reductions in median CD19+ counts were already observed at the first post-dosing assessment (after about 48 hours). Most of the patients who achieved the target depletion level started to replete at 43–341 days (supporting a monthly SC dosing) and reached the repletion criterion at 113–657 days. All B-cell subsets (naïve, naïve-mature and memory B cells) were depleted following ofatumumab dosing, with memory B cells (CD27+IgD-) reduced for a prolonged period of time in most depleted patients and repletion that was characterised by emergence of mainly naïve phenotype (CD27-IgD+). Median levels of BAFF increased in the B-cell-depleted dose groups, consistent with its functions as a B-cell survival factor.42 This study in patients with RA also demonstrated a good tolerability of SC ofatumumab at doses up to 60 mg with the use of oral antihistamine and acetaminophen premedication, without oral or intravenous glucocorticoids. Systemic injection reactions were mostly mild or moderate in intensity and manageable with additional oral acetaminophen or antihistamine.42
As a fully-human antibody, ofatumumab is predicted to be less immunogenic than rituximab, which is a chimeric mAb. This has been confirmed by the very low incidence (<1%) of anti-drug antibodies against ofatumumab observed in oncology studies.43 A positive anti-drug antibody response had no detectable impact on the safety, PK or PD of ofatumumab.
Phase II clinical trials in multiple sclerosis
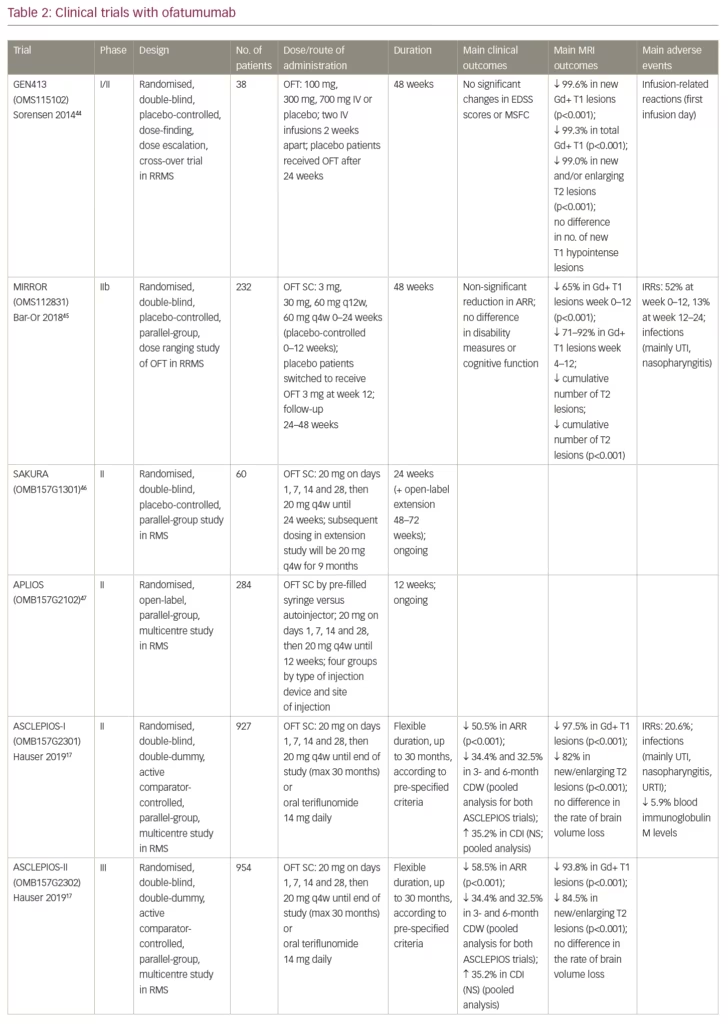
The safety and preliminary efficacy of intravenous ofatumumab were evaluated in a small phase I/II, 48-week, randomised, double-blind, placebo-controlled, cross-over (at week 24) study (ClinicalTrials.gov identifier: NCT00640328) in 38 patients with RRMS who were randomised to receive two infusions of ofatumumab (100 mg, 300 mg or 700 mg) or placebo 2 weeks apart (Table 2).17,44–8 Treatment with ofatumumab was well tolerated and was not associated with any unexpected safety concerns. Infusion-related reactions, mostly grade 1 or 2, were common on the first infusion day, but not observed on the second infusion day. The proportion of patients with infections was similar between the ofatumumab and placebo treatment groups. Most infections were categorised as grade 1 and no opportunistic infections were observed.44 Profound reductions were observed in the number of new T1 gadolinium-enhancing (Gd+) lesions, total number of T1 Gd+ lesions, and new and/or enlarging T2 lesions for all ofatumumab groups compared with placebo (p<0.001), but no significant change was observed in T1 hypointense lesions. These results should be interpreted with caution due to the small number of patients, and because one patient who was randomised to placebo had 88 T1 Gd+ lesions which accounted for around 75% of all the new Gd+ lesions at week 24. Relapses occurred in 19% of ofatumumab-treated patients and in 25% of placebo-treated patients and no significant changes in expanded disability status scale (EDSS) or MS functional composite scores were detected; however, this small study was not powered to detect any clinical efficacy. In addition, a less restrictive relapse definition used in this study may account for more relapses than in studies using more rigorous definition of a qualifying relapse (e.g., Sorensen et al.49). Similar to rituximab and ocrelizumab, ofatumumab treatment was associated with profound selective reduction of B cells as measured by CD19+ cells in circulation. B-cell counts decreased to zero within 1 week, and recovery started for the majority of patients in the 100 mg and 300 mg active/placebo groups after approximately 12 and 20 weeks, respectively, and continued during the second treatment period (week 24–48). Repletion was observed in a few patients in each dose group by week 48. No patients in this study tested positive for anti-drug antibodies, supporting the low immunogenicity of this human mAb.44
Following the development of a SC formulation of ofatumumab, the MIRROR trial (ClinicalTrials.gov Identifier: NCT01457924) was conducted.45 This was a phase IIb, 48-week, double-blind, placebo-controlled, parallel-group, dose-ranging trial that randomised 232 patients with active RRMS to SC ofatumumab 3 mg, 30 mg, or 60 mg every 12 weeks; ofatumumab 60 mg every 4 weeks for 24 weeks; or placebo followed by SC ofatumumab 3 mg at week 12. Patients in the 30 mg and 60 mg ofatumumab-dose groups were randomised to receive (in a 1:1 ratio) either placebo or a conditioning dose of ofatumumab (3 mg) 1 week prior to receiving their first active treatment dose, in order to evaluate whether tolerability to the higher ofatumumab could be enhanced by administration of an initial, smaller dose of ofatumumab (which may provide for more gradual lysis of B cells and potentially reduce cytokine release reactions [Table 2]). The mean rate of cumulative new Gd+ brain lesions over 12 weeks (the primary end point) was reduced by 65% for all dose regimens versus placebo (p<0.001), and by 71–92% between weeks 4–12 in a dose-dependent manner, with ≥90% suppression of new lesions at all cumulative doses ≥30 mg over 12 weeks. Similar dose-dependent reductions in the cumulative number of new Gd+ lesions were observed at week 24 and 48, too. All secondary MRI endpoints, including the cumulative number of T2 lesions, supported the primary analysis. Rapid dose-dependent CD19+ B-cell depletion was observed, which correlated with efficacy outcomes. However, complete depletion was not necessary for a robust treatment effect, as the lowest dose of ofatumumab (3 mg) every 12 weeks reduced circulating B-cell levels to ~25% of baseline (clearly less than the 5% of baseline achieved with the 30- and 60-mg dose every 12 weeks), while it was surprisingly effective in significantly reducing new T1 Gd+ lesions (but still allowing for disease activity to persist). The reduction of Gd+ lesions was related to the number of circulating B cells, with the strongest effect on MRI lesion activity observed at a depletion level of ≤8 cells/µL. B-cell repletion was achieved by 64–74% of patients by study end, with longer time to onset of repopulation for the higher-dose groups, and faster in all ofatumumab doses than previously reported with other anti-CD20 therapies. This dose response in the kinetics of depletion–repletion suggests that higher-dose/higher-frequency regimens result in greater depth of B-cell depletion in tissues, as observed also in animal studies.50 Over the first 24-week period, 17 (25%) patients relapsed in the placebo group versus 3–10 patients (9–22%) across the ofatumumab groups. The proportion of relapses remained low throughout the 24-week follow-up phase across all dose groups (6–15%). There were no significant differences between ofatumumab and placebo in measures of disability and fatigue, and EDSS scores remained unchanged in most patients.45
Adverse events (AEs) were largely mild-to-moderate in severity, and no patient died. The most common AE was injection-related reactions (IRRs) of mild-to-moderate severity in 97%, most commonly associated with the first dose and diminishing on subsequent dosing. Similarly to the OMS115102 trial with intravenous ofatumumab,44 overall rates of any infection-related AEs were similar across treatment groups, with no cases of opportunistic infections. AEs leading to withdrawal were reported in ≤2% of patients in each phase of the trial. In total, eight patients discontinued treatment because of AEs, mostly IRRs (two patients) and decreased IgG levels (two patients).45 Four patients had a single positive result for anti-drug antibodies during the treatment phase (all titers ≤32), and one patient also had a positive titer during follow-up phase week 36 (negative at week 48).45
Overall, this study demonstrated that: 1. ofatumumab is highly effective in suppressing new brain inflammatory activity with SC administration in a dose-dependent manner and at considerably lower doses compared with those previously studied in patients with MS; 2. B-cell repletion between dosing occurs faster than previously reported with other anti-CD20 therapies and is seen with the less frequent administration of ofatumumab (i.e., every 12 weeks), but not with the more frequent dosing (i.e., every 4 weeks), supporting a monthly administration; 3. complete depletion is not necessary for a robust treatment effect; 4. as a fully-human mAb, ofatumumab exhibits, as expected, very low immunogenicity; and 5. SC administration of ofatumumab is associated with good tolerability and no new/unexpected safety findings, which may enable more practical and convenient self-administration of ofatumumab than intravenous administration and reduced utilisation of healthcare resources. These findings supported the design of phase III studies in MS using SC injections of ofatumumab 20 mg administered every 4 weeks.
Another phase II study (ClinicalTrials.gov Identifier: NCT03249714) is a 24-week, randomised, double-blind, placebo controlled, parallel-group, multicentre study that evaluates the efficacy, safety and tolerability and PK of ofatumumab versus placebo (2:1 ratio) in 60 patients with relapsing MS, which is currently being conducted in Japan and Russia.46 The study has an extension, in which all patients receive open-label ofatumumab for an additional 24–48 weeks. The primary endpoint is the cumulative number of Gd+ T1 lesions across four MRI scans at weeks 12, 16, 20 and 24. In order to demonstrate PK bioequivalence of SC ofatumumab injected by pre-filled syringe used in clinical trials and the to-be marketed autoinjector, another randomised-open label study named APLIOS (ClinicalTrials.gov Identifier: NCT03560739) is currently being conducted in 284 patients with active RMS.47 The primary endpoints are area under plasma concentration-time curve over dosing interval (AUCtau) and the maximum observed plasma concentration (Cmax). Other objectives include the characterisation of the PK following SC administration of ofatumumab to either the abdominal region or the thigh and assessment of immunogenicity during the 12 weeks duration of the study addressing potential differences in ofatumumab anti-drug antibody formation between the pre-filled syringe and autoinjector devices as well as between abdomen and thigh injection sites.
Phase III clinical trials in multiple sclerosis
Following the success of phase II studies, two identical independent phase III clinical trials (ASCLEPIOS I, NCT02792218 and ASCLEPIOS II, NCT02792231) have been conducted globally. These were double-blind, double-dummy, active comparator-controlled, parallel-group, innovative, adaptive design, multicentre trials that randomised (1:1) 927 and 954 patients with active relapsing MS, respectively, to receive either SC ofatumumab 20 mg every 4 weeks (after an initial loading regimen of 20 mg SC doses on days 1, 7 and 14), or teriflunomide 14 mg orally once daily, for up to 30 months. The studies had flexible durations, with termination occurring in the blinded core treatment epoch according to pre-specified criteria. Patients aged 18–55 years with EDSS scores of 0–5.5 at screening who experienced ≥1 relapse in the past year, or ≥2 relapses in the past 2 years, or had a positive Gd+ MRI scan during the year before randomisation, were included. The primary endpoint was the annualised relapse rate. Key secondary endpoints included 3- and 6-month confirmed disability worsening (CDW), 6-month confirmed disability improvement (CDI), MRI-related outcomes and serum neurofilament light chain levels. Safety and tolerability have also been assessed. Results from these studies have recently been reported at the ECTRIMS (European Committee for Treatment and Research in Multiple Sclerosis) congress in Stockholm in September 2019,17 but have not been published yet in a peer-reviewed journal by the time of submitting this manuscript.
At the end of the trials, patients receiving ofatumumab had significantly lower annualised relapse rates than patients receiving teriflunomide (0.11 versus 0.22 and 0.10 versus 0.25 in the ASCLEPIOS I and II studies, respectively, p<0.001) (Table 2). Pooled analysis showed significant reductions in 3-month (p=0.002) and 6-month (p=0.012) CDW, but the 32.5% increase in the number of patients experiencing 6-month CDI with ofatumumab did not reach statistical significance. Treatment with ofatumumab was associated with a profound decrease in the number of Gd+ T1 brain lesions and significant reductions in the number of new or enlarging T2 lesions on brain MRI (p<0.001 for all) (Table 2). There was a significant decline in the levels of serum neurofilament light-chain (NfL) in the ofatumumab-treated patients compared with the teriflunomide-treated patients beginning at month 3 and reaching 23–24% difference (p<0.001), with almost no fall in the comparator group, suggesting strong decrease in ongoing neuronal damage by ofatumumab. On the other hand, there was no significant difference between groups in the rate of brain volume loss (BVL) (Table 2). It should be noted, however, that teriflunomide by itself is effective in reducing the rate of BVL in MS.51 AEs were balanced between groups and the safety profile of ofatumumab was quite favourable. There was a slight increase in IRRs (mild-to-moderate in 99%) and injection-site reactions in the ofatumumab group, but the rate of common infections was similar and no opportunistic infections were observed. Blood levels of IgM were reduced in 5.9% of the patients treated with ofatumumab. Serious AEs (SAEs) were reported in 9.1% in the ofatumumab group and in 7.9% in the teriflunomide group, including five and four malignancies, respectively. Two malignancies were pre-existing in the ofatumumab group. It should be emphasised that these are preliminary results presented at a scientific meeting after an initial analysis that may differ from the final published results.
Patients who completed their participation in one of the MS trials with ofatumumab are offered the opportunity to continue treatment in the ALITHIOS open-label trial (ClinicalTrials.gov Identifier: NCT03650114) aimed at collecting long-term safety, tolerability, effectiveness and health-outcomes data, and finding out the effects of ofatumumab on the development of antibody responses to selected vaccines and to the keyhole limpet hemocyanin (KLH) neo-antigen.48
In summary, ASCLEPIOS I and II studies, in a broad RMS population, demonstrated that ofatumumab 20 mg administered by SC injections once a month (versus teriflunomide) showed superior efficacy in lowering relapse rate and MRI activity, substantial and significant reductions in 3- and 6-month CDW, lower levels of NfL already present at month 3 and a favourable safety profile with no unexpected safety signals.
How do ofatumumab and ocrelizumab compare?
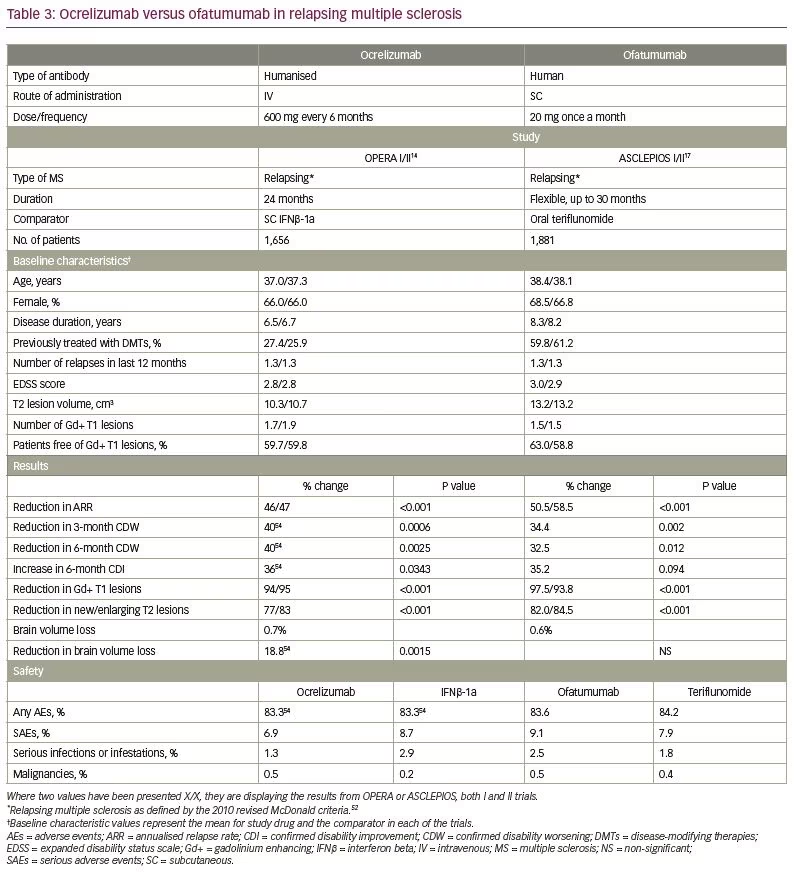
Direct comparison across clinical trials is difficult because of differences in trial design, study populations, selection criteria, endpoints, definitions (e.g., diagnostic criteria, relapses) and statistical analyses. Nevertheless, it may be of interest to compare data from the phase III clinical trials with ofatumumab and ocrelizumab, a humanised anti-CD20 mAb administered intravenously every 6 months and approved for both relapsing and PPMS.14 As shown in Table 3,14,17,52,54 most baseline characteristics in the OPERA I and II trials with ocrelizumab and in the ASCLEPIOS I and II trials with ofatumumab were comparable. However, patients in the ofatumumab trials were a little older and had longer disease duration than patients in the ocrelizumab trials, which may indicate a more advanced disease. Furthermore, most patients in the OPERA trials (73.4%), compared with <40% in the ASCLEPIOS trials were treatment naïve, where a better response to treatment is expected. Nevertheless, clinical and MRI outcomes were quite similar for both drugs, with the exception of 6-month CDI and the effect on BVL.
The difference in the proportion of patients with 6-month CDI was similar for ofatumumab versus teriflunomide (35.2%) and ocrelizumab versus SC IFNβ-1a (36%). However, these differences translated only to a trend for ofatumumab to increase CDI (p=0.094), and a statistically significant effect for ocrelizumab (p=0.0343). This difference can be due to the smaller number of patients achieving CDI with ofatumumab versus ocrelizumab (11% versus 15.6%, respectively), which further supports differences in baseline characteristics between the clinical trials that may favour a better response to ocrelizumab.
Although the magnitude of BVL was similar for ofatumumab (0.7%) and ocrelizumab (0.6%), only ocrelizumab significantly reduced the rate of BVL compared with the comparator (SC IFNβ-1a). A possible explanation is the ability of the comparator (teriflunomide) in the ofatumumab trials, by itself, to slow down BVL in MS.51 In addition, the method used to measure brain volume, which was not yet reported for the ASCLEPIOS trials, may also impact on brain volume measures. An example is the TEMSO trial, where initial calculations of the brain parenchymal fraction did not show any effect of teriflunomide on the rate of BVL,53 while later analysis using the SIENA method revealed a significant reduction of BVL versus placebo that was achieved with teriflunomide.51
The rates and types of AEs and SAEs also look similar for ocrelizumab and ofatumumab.14,17,54 Nevertheless, more time is required to appreciate the long-term safety profile of ofatumumab in MS. Although rates of infections were similar in the pivotal clinical trials for ofatumumab and teriflunomide, as for ocrelizumab and IFNβ-1a, and no opportunistic infections were reported, updated safety analysis from all clinical trials with ocrelizumab performed in 2019 revealed some increase in the rate of serious infections over time and six cases of potential serious opportunistic infections.55 By that time, seven cases of progressive multifocal leukoencephalopathy (PML) were reported in patients with MS treated with ocrelizumab, all carried over from prior treatments.56 However, a first case of de novo PML in a 78-year-old patient with MS treated with ocrelizumab as a first-line therapy has recently been reported,57 raising a question about the safety of initiating cell-depleting therapies that might accelerate immunosenescence in elderly patients. PML has previously been described also in patients treated with ofatumumab for B-cell haematological malignancies,58 and is not unexpected in MS, too.
Conclusions and future directions
Targeting of the B-cell surface antigen CD20 by mAbs has been shown to be effective in the treatment of both relapsing and progressive forms of MS.59,60 This journey started with rituximab, a chimeric anti-CD20 mAb that provided a strong proof-of-concept of the important role of B cells and anti-B-cell therapy in MS; moved on to ocrelizumab, a humanised mAb that showed high efficacy in suppressing inflammatory disease activity in MS and is currently approved for relapsing forms of MS (including active SPMS), and for PPMS; and now we are looking forward to the upcoming approval of ofatumumab for relapsing forms of MS. Ofatumumab is suitable for a first-line treatment for patients with active relapsing MS and may have several advantages over other anti-CD20 mAbs: it is a fully-human antibody exhibiting a very low immunogenicity and possessing several characteristics that may increase its efficacy in targeting B cells. Moreover, the SC administration of ofatumumab is simpler and more convenient than the intravenous administration of other anti-CD20 mAbs, allowing for self-administration at home and savings in health resource (e.g., infusion centres) utilisation.
Several open questions still remain about the place of ofatumumab in the treatment of MS: is it also effective in active PPMS like ocrelizumab? What will be its long-term safety? Shall we see increasing rates of infections and malignancies in the future? Is there an issue with reduced immunoglobulin levels? With the availability of several B-cell-depleting agents, which patients will respond best to ofatumumab? How long should a patient with MS be depleted of peripheral B cells with monthly injections that do not allow for B-cell repletion? Is complete B-cell depletion required for optimal treatment response, or may partial depletion be sufficient? Is there a place for future strategy of combining another B-cell-depleting agent that can cross the blood–brain barrier and target compartmentalised inflammation in the CNS that contributes to disease progression? More clinical and observational studies and long-term surveillance are required to answer these questions.
Advances in mAb technology have also resulted in the development of third-generation mAbs such as ublituximab, a novel chimeric glyco-engineered IgG1 mAb that binds a unique epitope on the CD20 molecule and demonstrates increased binding capacity to CD20 and increased ADCC activity due to defucosylation of its Fc region, which increases the affinity for FcγRIIIa, resulting in more efficient immune effector cell engagement and enhanced target cell killing.61 Following a successful phase II study,16 ublituximab is currently being studied in two identical phase III ULTIMATE trials and may join the increasing armamentarium of anti-CD20 B-cell-depleting agents in the future. Another attractive approach to target B cells is by using inhibitors of Bruton’s tyrosine kinase (BTKi). These small molecules inhibit B cells as well as myeloid cells, can cross the blood–brain barrier and potentially target B cells and activated microglia within the CNS, both of which contribute to disease progression in MS. Several phase II and III studies are currently ongoing or planned with a variety of BTKis in all types of MS, including a completed phase II trial with evobrutinib in relapsing MS.62 BTKis may potentially be added to B-cell-depleting mAbs such as ofatumumab as a possible future strategy to efficiently target B cells both in the periphery and in the CNS.
In summary, the fully-human anti-CD20, B-cell-depleting mAb ofatumumab has shown high efficacy, good tolerability and a favourable safety profile in patients with relapsing MS. Its approval, which is expected soon, will make a considerable contribution to the expanding arsenal of effective biological therapies for MS.