The clinical use of cancer immunotherapy with immune checkpoint inhibitors (ICI) has transformed cancer management and added another effective treatment option for different types of malignancies.1–3 In 2018, the Nobel Prize for medicine and physiology was awarded for the discovery of immune checkpoint molecules. Therapy with ICI has improved patients’ survival and quality of life and has enabled more effective treatment of previously refractory cancer. Treatment protocols have utilized ICI as monotherapy, dual therapy (two different ICIs), or in combination with chemotherapy, targeted therapies or other cancer treatment modalities. While the high cost of ICI limits their availability as a treatment, the growing number of indications and the increased use outside of academic centres have greatly expanded their use. In 2018, it was estimated that 44.0% of cancer patients may be eligible for treatment, and 13.0% may respond to treatment with ICI.4
Immune checkpoint molecules serve as co-inhibitory molecules and represent an important regulatory mechanism in the immune system for maintaining immune homeostasis and promoting self-tolerance.3 Immune checkpoint molecules and their ligands are primarily expressed by dendritic cells, T cells and B cells, but the expression of immune checkpoint molecules by tumour cells may limit the immune response.3 Conversely, the blockade of immune checkpoint molecules enhances the immune response and may suppress tumour growth and metastases.1,3 There are multiple immune checkpoint pathways, and ICI in current clinical use target cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed cell death-1 (PD-1)/ligand 1 (PD-L1) pathways.
The first ICI in clinical use was a monoclonal antibody targeting CTLA4, ipilimumab (YERVOY®; Bristol Myers Squibb, New York, NY, USA), which was approved in 2011 for the treatment of metastatic melanoma (Table 1).5 Three years later, two antibodies targeting PD1 – nivolumab (OPDIVO®; Bristol Myers Squibb, New York, NY, USA)6 and pembrolizumab (KEYTRUDA®; Merck & Co., Inc., Kenilworth, NJ, USA)7 – were first approved for the treatment of advanced melanoma and later for the treatment of non-small cell lung cancer, renal cell carcinoma and lymphoma (Table 1). Other treatments targeting malignancies such as CTLA4, PD-1 and PD-L1 have followed (Table 1).

However, an enhanced immune response may also lead to autoimmune complications or immune-related adverse events (irAEs), which may involve skin, endocrine, respiratory, gastrointestinal or neurologic manifestations.8,9 Prevalence of irAEs of any grade of severity after ICI was estimated at 20.0–60.0%, with an ICI-related mortality rate of 0.3–1.3% among the treated patients.9 The use of ICI has led to long-term remissions in some patients who previously had a terminal disease, and the presence of irAEs often correlates with improved tumour response.10 Pathophysiology of irAEs includes T cell activation, antibody production and cytokine release.9 Combination immunotherapy with CTLA4 and PD 1/PD L1 inhibitors is even more effective in treating different cancers than monotherapy; however, it is also associated with a greater risk of autoimmune complications. As the indications have expanded, ICI has evolved beyond rescue therapy or refractory malignancies and may now represent a first-line option for some types of cancer. Therefore, ICI and its complications are now increasingly encountered in the daily clinical practice of primary care physicians, neurologists and other non-oncology specialists. Neurologic complications of ICI usually manifest during treatment, within the first 3 months from treatment onset, but delayed complications may occur as well.9 Post-ICI immune response is not target specific and may lead to increased titers of various antibodies. Rarely, post-ICI irAEs can be associated with elevated titers of neurologic autoantibodies, including novel ones that have not been previously reported.11 Neurologic complications are less common than endocrinologic, gastrointestinal and skin irAEs; this discrepancy may be attributable to the immunologically privileged status of the nervous system, for example, because of the role played by the blood–brain and the blood–nerve barriers.12 It has been estimated that neurologic complications may affect up to 2.0–4.0% of patients treated with ICI, with clinically relevant and severe neurotoxicity affecting fewer than 1.0% of patients.13–15 Neurologic irAEs may present de novo or manifest as exacerbations of pre-existing neurologic disorders. Multisystemic irAEs, including gastrointestinal, skin, pulmonary, endocrine or neurologic irAEs, are fairly common after ICI treatment.14,16,17 While different types of irAE are not specific to specific cancer types or ICI treatments, the frequency of individual irAEs is influenced by ICI and the type of cancer. The severity of post-ICI neurologic complications varies broadly from mild symptoms not requiring a change of therapy to rapidly progressive and even fatal cases. Historically, clinical trials with ICI have excluded patients with previous autoimmune disorders, as these patients may be at greater risk of irAEs. However, these patients are now eligible for treatment with ICI in clinical practice, and it has been estimated that about 50.0% of such patients with autoimmune disorders may complete ICI treatment without significant autoimmune complications.18
In addition to cancer immunotherapy-induced iatrogenic immune checkpoint blockade, immune checkpoint molecule deficiency was described with various autoimmune conditions, including giant cell arteritis, in which the breakdown of the tissue-protective PD-1/PD-L1 checkpoint may promote vascular inflammation.19
Neurologic complications of immune checkpoint inhibition
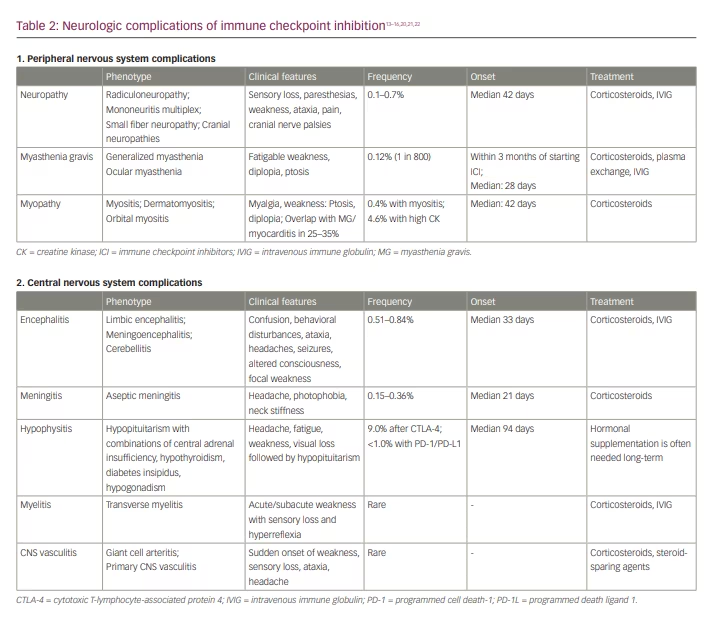
The clinical manifestations of immune checkpoint inhibition neurotoxicity may affect both the central nervous system (CNS) and the peripheral nervous system (PNS) (Table 2).13–16,20,21,22 Neurologic irAE after ICI may occur at any time, but neuromuscular complications generally manifest earlier (median: 70 days) than CNS complications (median: 119 days).20
Immune response associated with neurologic irAEs involves T cell activation, cytokine release and autoantibody production, both with previously described and novel autoantibodies.14,23–26 Rarely, neurologic syndromes associated with elevated titers of paraneoplastic neuronal antibodies involving both the PNS and the CNS have been reported after ICI.20,22 High rates of autoantibodies are found with post-ICI myasthenia, similarly to non-ICI myasthenia, and patients with myasthenia/myositis/myocarditis overlap may have elevated titers of antibodies targeting acetylcholine receptors, striated muscle antibodies and myositis-specific antibodies (MSA).27 In contrast to post-ICI myasthenia, most series describe the low prevalence of MSA in patients with post-ICI myositis without overlap syndromes.28,29 Novel autoantibodies have been described in patients with various neurologic irAEs, including myasthenia gravis, polyradiculoneuropathy and myelitis.24 In an animal model of myasthenia gravis, CTLA-4 blockade led to epitopal spreading and enhanced immune response.30 Additionally, an animal model blockade of CTLA4 also demonstrated a paraneoplastic-like syndrome analogous to paraneoplastic cerebellar degeneration.31 Conversely, ongoing immunosuppression of autoimmune neurologic disorders may limit anti-tumour immune response and potentially reduce the benefits of ICI, such as fingolimod potentially reducing the number of tumour-infiltrating lymphocytes after ICI.32
Neurologic complications of the central nervous system
The neurologic complications of ICI may manifest as encephalitis, cerebellitis, meningitis, CNS vasculitis, hypophysitis, neurosarcoidosis or multiple sclerosis and related syndromes.14,33–36 A greater risk of CNS complications after ICI was reported in patients with non-small cell lung cancer.33 Immune-related ICI complications of the CNS are, to a great extent, a ‘diagnosis of exclusion’; therefore, diagnosing these complications is valuable, as an appropriate workup should exclude other potential causes of symptoms.
Encephalitis associated with immune check inhibitors may present with various phenotypes, including limbic encephalitis and meningoencephalitis. Serology pursued in the workup of encephalitis is often negative, although some patients may have elevated titers of anti-neuronal autoantibodies.25,37,38 Like patients with non-ICI limbic encephalitis, patients with post-ICI limbic encephalitis often present with altered mental status, seizures and psychiatric symptoms.33 Limbic encephalitis may manifest with new onset of psychiatric symptoms after treatment with ICI, and it should not be mistaken for a neurocognitive disorder. Meningoencephalitis may manifest with altered mental status, fever, memory disturbances and headaches. There may be an overlap between CNS and PNS complications.
Some patients may also develop cerebellitis with gait ataxia, limb ataxia, opsoclonus–myoclonus syndrome and/or cranial neuropathies.33 Aseptic meningitis manifests with neck stiffness, fever, and headache and needs to be differentiated from infectious meningitis or carcinomatous/lymphomatous leptomeningeal spread.
The most common symptoms of hypophysitis are headaches and visual loss. Consequently, although this condition is often listed as an endocrine complication, a neurologist may be the first specialist to evaluate patients with hypophysitis. Other common symptoms of hypophysitis include severe fatigue, hypotension and nausea.21 Interestingly, higher rates of post-ICI hypophysitis have been reported among men, contrasting with the higher prevalence of other ICI-related endocrinopathies in women.39 Similarly to ‘idiopathic’ pituitary apoplexy, a typical clinical manifestation of hypophysitis may include an acute onset with severe headache and loss of vision followed by endocrinopathy or may present more insidiously.40 Hypophysitis and hypopituitarism are much more common in patients who have received ICI treatment than in the general population and may affect up to 9.0% of patients after treatment with CTLA-4 inhibitors.21,41 In addition to the effects of an enhanced immune response, as with other irAE, there may also be a direct effect of anti-CTLA-4 antibodies on the pituitary gland, as CTLA-4 is expressed in some pituitary cell types. Treatment with corticosteroids may not be effective, as the diagnosis is usually made only after a pituitary injury has already occurred, and subsequent endocrine supplementation affecting multiple hormonal axes may be needed.40
Few cases of post-ICI systemic and single-organ vasculitis, including giant cell arteritis and primary angiitis of the CNS, have been described.42 Discontinuation of ICI and treatment with corticosteroids have resulted in the resolution of symptoms in most cases.42
Furthermore, multiple sclerosis and related syndromes, including optic neuritis and transverse myelitis, may develop de novo or relapse after immune checkpoint inhibition.35,36,43 Exacerbations of multiple sclerosis are more common than reports of de novo onset. De novo onset of multiple sclerosis may also represent the unmasking of subclinical or previously asymptomatic CNS demyelination. Optic neuritis is often bilateral and may be associated with uveitis.44 Atypical demyelinating lesions may resemble progressive brain metastases and may resolve spontaneously after ICI is stopped.45 Post-ICI myelitis may present in combination with other complications, including posterior reversible encephalopathy syndrome and hypopituitarism, and most patients improve with treatment.46
Finally, multisystemic sarcoidosis involving the CNS or PNS and other organs and tissues may develop (or get unmasked) following immune checkpoint inhibition.47
Neurologic complications of the peripheral nervous system
Neuromuscular complications of immune checkpoint inhibition include myopathy with a wide spectrum of phenotypes, myasthenia gravis and different variants of peripheral neuropathy. Complex overlap syndromes with various combinations of myasthenia gravis, myositis and inflammatory neuropathies are much more frequent after ICI compared with non-ICI neuromuscular disorders.14,26
The clinical presentation of post-ICI myositis typically manifests with myalgias, proximal weakness, and fatigue and is mostly indistinguishable from non-ICI inflammatory myopathies. Similarly to other irAEs, post-ICI myositis is driven by the loss of immune tolerance, and some of the proposed pathophysiologic mechanisms associated include regulatory T cells (Treg) dysregulation, epitope spreading and sharing, direct toxicity and pre-existing auto-immunity.48 Inflammatory myopathy associated with ICI often has a similar histopathology to immune necrotizing myopathy (IMNM) or dermatomyositis.28,49–51 Serum creatine kinase (CK) levels may be as high as 16,620 units per litre (U/L); however, compared with IMNM, the median CK is typically much lower (686 versus 6,456 U/L).28,52 The incidence of post-ICI myositis has been estimated at 0.4%, and up to 4.6% of patients develop hyperCKemia after ICI, which is usually asymptomatic.53,54 While post-ICI myopathy may be associated with significant morbidity, most patients with post-ICI hyperCKemia remain asymptomatic, and routine CK screening or monitoring after ICI use does not provide significant clinical benefit.53 Unusual post-ICI myopathy presentations may include axial and oculomotor symptoms.52 Greater risk of hyperCKemia after ICI was reported with incidental statin use, but the clinical significance of this finding remains uncertain.55 Immune adverse effects of ICI may also unmask underlying hereditary neuromuscular disorders, as has been described with statin myotoxicity.56,57
Myositis specific autoantibodies are usually absent in patients with ICI myopathy, but striated muscle antibody titers may be elevated in up to 68.0% of patients.28,29 Some patients may develop myocarditis and/or orbital myositis.58 Myocarditis after ICI is associated with significant morbidity and mortality and may manifest with asymptomatic abnormalities of cardiac biomarkers, chest pain, arrhythmias or even cardiogenic shock.51,58
Post-ICI orbital myositis may present with ptosis or diplopia mimicking ocular myasthenia gravis, with orbital MRI showing enhancement of orbital muscles.58 Myasthenia gravis has been reported in 0.12% of patients treated with ICI (1 in 800), compared with 1 in 12,000 in the general population.60 Myasthenia typically presents as acetylcholine receptor seropositive generalized myasthenia of varying severity and almost always starts within 3 months of starting ICI.21,61 De novo myasthenia gravis is more common, with exacerbations reported in 23% of post-ICI MG cases, in contrast with the rare occurrence of de novo multiple sclerosis after ICI.26,35 While the initial case series of post-ICI myasthenia gravis did not include cases associated with elevated anti-muscle-specific kinase antibodies, more recent studies suggest that it may occur with a similar frequency as in idiopathic myasthenia gravis.26,62
Post-ICI neuropathies manifest with a wide spectrum of phenotypes, including Guillain-Barre-like syndrome (GBS-like) with or without cranial nerve involvement, mononeuritis multiplex, axonal or demyelinating polyneuropathy, plexopathies, radiculoneuropathies, cranial neuropathies, and small fibre neuropathy with or without dysautonomia.16,26,63–66 In patients with radiculoneuropathy, MRI imaging may show enhancement of the spinal roots.18,66 Most patients improve with corticosteroids. Cerebrospinal fluid testing often shows pleocytosis and high protein content in patients with a post-ICI syndrome similar to GBS and polyradiculoneuropathy, which may improve with corticosteroids, unlike idiopathic Guillain Barre syndrome.16,67 Higher prevalence of paraneoplastic onconeural antibodies has been reported in neuropathy patients with neuroendocrine tumours after ICI compared with non-neuroendocrine tumours.68
Cranial neuropathies after ICI may be combined with CNS complications or presented as isolated events.33,63 Most frequently, facial, vestibulocochlear, optic and abducens nerves are involved.40,59 Similarly to CNS vasculitis, vasculitis of the PNS following ICI may manifest with painful vasculitic neuropathy.64,69
Evaluation
In the evaluation of cancer patients with new neurologic symptoms, after treating them with ICI, it is essential to determine whether these symptoms are related to the worsening of cancer or to cancer treatment, as this would also guide cancer treatment strategies. A careful history, examination and diagnostic testing may help establish whether recent neurologic events after use of ICI represent de novo neurologic complications, exacerbations of pre-existing neurologic disorders, complications from other (non-ICI) cancer therapies or further progression of the underlying malignancy.70,71 Multidisciplinary coordination of care is essential to maximize treatment benefits and reduce treatment-related morbidity, especially in diseases with multisystemic complications. Patients with a history of autoimmune disorders may be at higher risk of neurologic and non-neurologic irAEs.18 The improvement of neurologic symptoms after stoppage of ICI treatment and treatment with corticosteroids or other immunosuppressive medications supports the diagnosis of ICI-related neurologic events, although some patients may develop refractory or rapidly progressive neurologic complications that may not respond to therapy.70 In patients with suspected post-ICI myocarditis, electrocardiogram may be normal, despite having abnormal troponin and CK myocardial band levels, but cardiac MRI or myocardial biopsy may indicate myocarditis, so a prompt cardiology consultation is essential.52,59 High-risk patients with multiple comorbidities and a history of autoimmune diseases should be monitored for treatment-related complications. Educating patients and healthcare providers may also facilitate early recognition of various complications and reduce associated morbidity.72
Treatment
The treatment of adverse events associated with cancer immunotherapy needs to balance the risks of cancer progression versus the complications of adverse autoimmune events. Detailed guidelines have been provided in the literature describing the stepwise approach to evaluating and treating patients with post-ICI complications and irAEs.70,71,73 Mild irAE may warrant only clinical monitoring. In patients with more severe irAEs, withdrawing ICI may be effective for irAE (Figure 1) but can also lead to the progression of the underlying cancer. The decision to discontinue ICI is based on the severity of the autoimmune complications and circumstances of cancer treatment, considering the specific clinical needs of the individual patient. Typically, corticosteroids are the backbone of most treatment protocols and the first medication tried to treat irAEs. Corticosteroids may also be combined with immunomodulatory (intravenous immune globulin, plasma exchange) or immunosuppressive (e.g. mycophenolate, azathioprine, rituximab) treatments. The use of aggressive immunotherapies for irAEs may potentially reduce the efficacy of cancer treatment, and such decisions should be made carefully on a case-by-case basis.
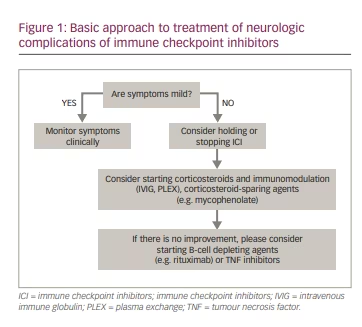
Another important issue is the risk of recurrence of immune checkpoint inhibition neurotoxicity after re-challenge with the same or a different type of ICI, which may be cautiously pursued in most patients.15,71,74
Summary
Immune checkpoint inhibition presents a wide spectrum of neurologic complications, ranging from mild symptoms only requiring monitoring without intervention to severe neurologic complications warranting stoppage of cancer immunotherapy and immunosuppressive treatments with risks of severe morbidity and mortality. There is an increased risk of autoimmune ICI complications in patients with a history of different connective tissue diseases – including polymyositis/dermatomyositis, systemic lupus erythematosus and Sjögren’s syndrome – or vasculitis, and of myasthenia gravis, and these patients may require personalized surveillance of possible autoimmune complications. The risk is also greater when combining ICI of different classes. Post-ICI complications have to be differentiated from other cancer-related complications or cancer worsening. Typical treatment of post-ICI complications includes corticosteroids, possibly combined with immunomodulating and immunosuppressive therapies. Additionally, immune checkpoint molecules may play a role in the pathogenesis of different autoimmune neurologic disorders in the general population. In the evaluation of suspected neurologic complications of cancer immunotherapy, it is essential to exclude complications directly associated with cancer progression and complications of other cancer treatments. While the phenotypes of most individual neurologic complications are not significantly different from similar events in the general population, there is a high prevalence of overlap syndromes, with multiple autoimmune events occurring at the same time. One important overlap syndrome is the overlap of myasthenia gravis or myositis with myocarditis, as myocarditis can be associated with significant morbidity and mortality.
Accurate and timely diagnosis and treatment of post-ICI neurologic complications, including stopping cancer immunotherapy when indicated, is essential to improve outcomes and patients’ quality of life.