Autoantibody-mediated diseases of the central nervous system (CNS) are a rapidly expanding field within contemporary neuroimmunology. Autoimmune encephalitis (AIE) is a treatable cause of subacuteonset memory loss and confusion. In some patients, the evidence for inflammation is limited and the term autoimmune encephalopathy may be preferred. The associated spectrum of autoantibodies against specific molecules in the CNS is widening, and serologic testing can help reach a definitive diagnosis. Although each individual autoimmune encephalitis syndrome has some distinctive demographics and associated clinical features, an autoimmune cause for recent-onset amnesia, confusion, and seizures should be considered in men and women, adults and children alike. Importantly, AIE is often a treatable neurologic condition and hence its consideration and recognition is of great importance.
Autoantibodies can be divided into those that target the extracellular domains of membrane molecules and those against intracellular targets. While the former are believed to be pathogenic since they can access their targets on the membrane of live cells, the latter are likely to be markers of immune-mediated disease but unlikely to themselves be pathogenic.
Initially, AIE was considered a paraneoplastic phenomenon, typically associated with a number of tumors and many of the antibodies (Abs) targeted intracellular antigens (such as Hu, CV2/CRMP5 and Ma2). Patients with these antibodies and associated tumors are unlikely to respond to immunotherapy and have in general a poorer prognosis.1,2
More recently described antibodies associated with AIE have been directed against cell surface molecules and ion channels in particular. Ion channels are involved in crucial processes within the nervous system including direct signal transmission, synaptic plasticity, and neuronal network modulation. Any interruption of ion channel function can therefore be expected to impact on the performance and output of the CNS. By contrast to those with paraneoplastic encephalitis, patients withantibodies against surface antigens often have no underlying tumor and respond well to immunotherapy.3,4
A Glimpse at the Neuromuscular Junction
To understand how antibodies can cause disease, the neuromuscular junction (NMJ), and pathophysiology of myasthenia gravis (MG) provides a useful paradigm. Eighty percent of patients with MG have serum antibodies directed at nicotinic acetylcholine receptors (nAChR) located at the post-synaptic aspect of the NMJ. In individual patients, the reduction of antibody levels is accompanied by clinical improvement. The study of nAChR Abs has revealed a variety of different mechanisms of pathogenicity. Most Abs cross-link adjacent nAChR and lead to internalization and subsequent destruction of the receptor-Ab complex.5 Equally, many Abs lead to activation of the lytic cascade of the complement system resulting in local destruction of the NMJ.6 Abs against muscle specific kinase (MuSK), a molecule tightly associated with nAChRs, were subsequently found in seronegative patients with pronounced bulbar and facial muscle involvement.7 MuSK-MG has often a severe course that requires aggressive treatment. The identification of specific antigens was important in generating animal models which developed experimental autoimmune MG after they received the antibody or were immunized with the target antigen. Many of these principles are also true for the Lambert-Eaton myasthenic syndrome. This illustration of antibody-mediated disease in the peripheral nervous system2,5,6 illuminates some of the ways by which Abs can cause disease at a simple synapse and might offer some insight as to the potential complexity associated with studying their actions on central neuronal networks.
Autoimmune Encephalitides—An Evolving Spectrum of Antibody-associated Diseases
The subacute onset of memory loss, confusion, and seizures are characteristic features of AIE associated with neural antibodies. Viral encephalitis and metabolic conditions, such as herpes simplex encephalitis and Wernicke’s encephalopathy, respectively, can present with similar features. The clinical features can help make the diagnosis of an autoimmune process and then guide the rational identification of an associated Ab (see Table 1). Therefore, autoimmune encephalitides may be subdivided according to the antigen recognized by patient Abs.
As reviewed by Vincent and colleagues, the most common and well researched associated Abs are directed against the voltage-gated potassium channel-complex (VGKC-complex), N-methyl D-aspartate receptor (NMDAR) and glutamic acid decarboxylase (GAD).4 Small case series have also described patients with Abs against glycine receptors, amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), and gamma-aminobutyric B receptors (GABABR).
NMDAR-antibody Encephalitis
NMDAR-antibody encephalitis was first described as a paraneoplastic phenomenon in young females with an ovarian teratoma.8 The Ab target was then confirmed as the extracellular domain of the NR1 subunit of the NMDA receptor.9,10 In the following years, the clinical spectrum associated with NMDAR-Abs has widened significantly to include adult and pediatric, male and female, paraneoplastic and non-paraneoplastic patients.10 NMDAR-Ab encephalitis is now the most common AIE in younger patients and is a differential diagnosis to be considered in any patient presenting with subacute alterations of cognition, behavior and amnesia.11,12
Other consistently reported features of NMDAR-antibody encephalitis include seizures, dysautonomia, central hypoventilation, and sleep pattern alterations. However, the movement disorder is often the most distinctive single feature of the condition. This involves stereotyped, complex, prolonged orofacial, and limb movements which are often refractory to drugs usually used to treat chorea and dystonia. Most patients develop these varied symptoms within the first 4 weeks of their illness, often in a characteristic pattern.13,10
The majority of NMDAR-Abs are of the IgG1 subclass, however recently other NMDAR-Ab subclasses have also been reported. NMDAR IgM Abs were found in a patient with NMDAR-Ab encephalitis with features of bipolar disorder and the clinical improvement mirrored a fall in NMDAR IgM.14 In addition, 7/24 patients with a subacute decline of cognitive function were shown to have NMDAR IgA but not IgG and again, clinical improvement correlated with the serum levels of NMDAR IgA.15
Abs to the Voltage-gated Potassium Channel-complex
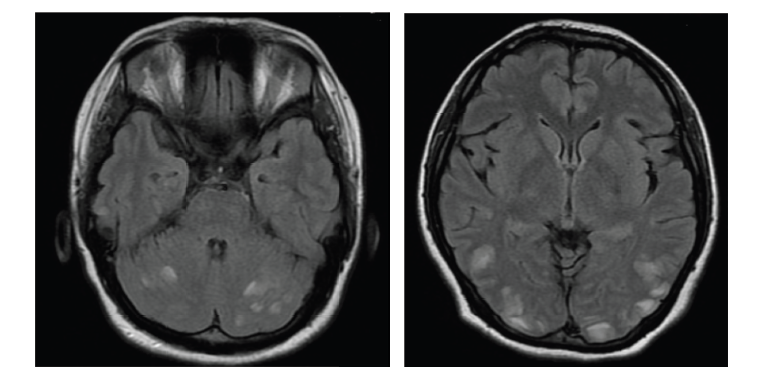
For over a decade, VGKC-complex Abs have been associated with clinically different syndromes: neuromyotonia (NMT16), Morvan’s syndrome,17–20 and LE.21–23 Patients with VGKC-complex Ab LE are more often male, older than 50 years of age and many respond well to immunotherapy. The majority of patients do not have tumors. The clinical features of VGKC-complex-Ab LE include subacute onset of amnesia, disorientation, neuropsychiatric changes, and seizures. Serum hyponatremia and signs of hippocampal inflammation on MRI are useful paraclinical pointers towards the diagnosis. Seizures are typically of medial temporal lobe semiology but it was recently reported that faciobrachial dystonic seizures (FBDS) are a frequent accompaniment. FBDS are brief (few seconds in duration), frequent (median 50 per day) events which typically affect the arm and face and precede VGKC-complex Ab LE in 75 % of cases.24–26
NMT is a peripheral nerve hyperexcitability condition that manifests with symptoms such as muscle cramps, stiffness, and fasciculations and shows an underlying thymoma in about 20 % of cases. Morvan’s syndrome describes the combination of NMT with CNS features. The latter are often with more psychiatric symptoms than in cases with LE and tumors (such as thymomas or small cell lung cancer) are found in about 40 % of cases.27,28
The diagnosis of VGKC-complex Abs relies on Ab-mediated precipitation of mammalian brain membranes that are radioactively labeled with alpha-dendrotoxin. The nature of the assay means that Abs against components closely associated with the VGKCs themselves will coimmunoprecipitate the radioactive-dendrotoxin-VGKCs. Further methods have to be used to identify the individual antigenic targets associated within the complex, and so far, three have been described: LGI1 (leucinerich glioma inactivated 1); CASPR2 (contactin associated protein 2); and Contactin-2.28,29 LGI1 was shown to be the target of the Abs found in patients with VGKC-complex Ab LE29,30 and also in cases with FBDS.25 By contrast, CASPR2-specific Abs are more commonly found in Morvan’s syndrome and NMT.31
Patients with Glutamic Acid Decarboxylase Abs
Patients with GAD Abs may present with a subacute onset of temporal lobe epilepsy (TLE) and cognitive impairment. Abs to the intracellular protein GAD are also present in other diseases such as stiff-person syndrome. Patients with GAD-Ab LE are often younger adult women (median age at onset = 47 years), mostly without a tumor. The disease has a progressive course, without phases of remission.32,33
Although GAD is an intracellular antigen and Abs against intracellular targets are believed less likely to be pathogenic per se, the typical changes seen on MRI in GAD LE patients are indicative of an inflammatory process within the temporal lobe. It is possible that as yet unidentified Abs against cell surface structures co-exist in the serum and are the actual pathogenic molecules driving the process. The patients’ response to oral immunotherapy is variable, but there are some successful reports of possible improvement following plasma exchange or intravenous immunoglobulin correlated with a reduction in Abs.34–36
Other Antigenic Targets
Other proteins involved in physiologic CNS synaptic processes have also been shown to be antigens in AIE. A small case series in 2009 identified 10 patients with LE and Abs against the GluR1 and GluR2 subunits of AMPAR.37 The patients were predominantly female (9/10) with an average age of 60 and the majority were also diagnosed with tumors (7/10). One year later, 15 LE patients were described with Abs against the GABABR.38 These patients were older, 62 years on average, mainly male (8/15) and 7/15 had lung tumors (5/7 were small cell lung cancer). In a further study of LE patients, 10/70 were shown to have GABABR-Abs, again the average age of the patients was 60 and 8/10 had small cell lung cancer.39 Antibodies against mGluR5 and DPPX have more recently been discovered.
Pediatric Autoimmune Encephalitis
Pediatric patients are an interesting emerging subgroup with AIE. Insults affecting the CNS may have more impact on the long-term outcome especially in very young patients where brain maturation and development is still a delicate ongoing process. Alternatively, children may have an intrinsically higher potential for plasticity. The two Abs that have been most commonly associated with pediatric AIE are NMDAR and VGKC-complex Abs.
In one study, NMDAR-Abs were reported in 32 pediatric patients and ovarian teratomas were present in one-third of the female patients under 18 and only one-tenth of those younger than 14 years.40 Many of the clinical features were similar to the adult cases. Subtle differences include a higher rate of prodromal infectious symptoms and the early presence of a movement disorder. The fact that pediatric cases are less likely to have tumors is relevant since a recent study has suggested the need for early aggressive treatment in non-paraneoplastic pediatric NMDAR-Ab encephalitis.41
The presentation of pediatric patients with LE and VGKC-complex-Abs appear to show some differences from the adult patient group. There are different rates of psychiatric/psychotic features, seizures, and infrequent reports of hyponatremia.42,43,44 Most of the reported pediatric cases are also female, whereas VGKC-complex-Abs are more common in male adult patients. Interestingly, many pediatric cases were VGKC-complex-Ab positive, without Abs against the known antigenic targets (LGI1, CASPR2, and contactin-2). 42 % of autoantibody positive (NMDAR or VGKC-complex) patients had a full recovery in a recently published case-series.43 A further case-series reported only 1/4 with complete recovery in pediatric VGKCcomplex autoantibody patients who presented with status epilepticus.45
Treatments for Patients with Autoimmune Encephalitis
Although no systematic studies of treatment regimens or RCTs for AIE have been undertaken, the available case series show a few patterns and overall, prompt immunotherapy and tumor management, when relevant, is believed to be key to improving patient outcomes.9,10,42,46,47
AIE can be subdivided into diseases with different antigens and these often show slightly different responses to treatment. It is therefore key to diagnose AIE promptly and exclude other causes for the patient’s presentation. The published case studies for all AIE associated with antibodies targeting extracellular domains of membrane proteins suggest that immunotherapies can help expedite recovery. As to which immunotherapy regimen is the best to follow, the verdict is still out, although recently studies have approached the question of patient management in NMDAR and VGKCcomplex Ab encephalitis. As VGKC-complex Abs are often associated with a monophasic illness, therapy is usually required for shorter periods and is less aggressive than in NMDAR-Ab encephalitis.
An observational cohort study summarized clinical findings in nearly 600 NMDAR patients treated at several centers. The authors suggested that 53 % of cases showed a good response to first-line immunotherapy (steroids, IVIg, PEx, alone or combined) within 1 month. Of the patients who did not respond to this treatment, the study concluded that longterm outcome was better if rituximab and/or cyclophosphamide were also administered.48 In 2004, VGKC-complex-Ab AIE was shown to respond to immunosuppressive treatments using steroids, IvIg or PEx with a fall in Abs levels and marked neuropsychologic improvement.46 The only published small prospective study in the field of VGKC-complex- Ab AIE showed favorable outcome in all nine VGKC patients where the protocol consisted of plasma exchange (PEx, 50 ml/kg), intravenous immunoglobulin (IVIg, 2 g/kg) and methylprednisolone (1 g x3), followed by maintenance oral prednisolone (1 mg/kg/day).47 Efficacy was also seen using monthly intravenous methylprednisolone pulses (500–1,000 mg/day on 3–5 consecutive days) which successfully improved seizure control (9/9 patients) and reduced VGKC-complex-Abs levels in 6/9 patients. However, the same regimen did not show a change in GAD levels (6/6 patients) or reduction in their seizure frequencies (0/7 patients).32 In a case series of patients with TLE and hippocampal sclerosis, 20/38 had previous LE or MRI changes consistent with LE emphasizing a potential role for autoimmunity in the development of TLE;49,50 maybe effective treatment regimes will delay or prevent the onset of this form of TLE. Antibody Detection and Pathogenic Mechanisms
A definitive diagnosis can be made using cell-based assays which depend on detection of bound patient IgG to the surface of HEK cells expressing the antigenic target at their plasma membrane. In general, patient serum has higher levels of Abs than CSF at onset and is sufficient for diagnosis; however, later on, or following failed treatments, CSF analysis may be helpful. Amongst the different AIE, most research into the pathogenicity of the Abs has been published in NMDAR-Ab encephalitis. Many of the mechanisms previously established in myasthenia gravis appear to be relevant to Absassociated with CNS pathologies (see Figure 1). Application of NMDAR-Abs can cause internalization of the NMDARs in vitro hippocampal neuronal cultures and in vivo after infusion of patient IgG into the hippocampus of rats.51 In cultures, NMDAR clusters disappeared 3 days after CSF incubation which was reversed when NMDAR-Abs were removed. NMDAR internalization can also be caused by NMDAR IgM or IgA Abs.14,15 In addition, NMDAR-Abs can show other potentially pathogenic actions—they may alter the trafficking and distribution of NR2A and 2B subunits around glutamatergic synapses (in some via an interaction between NMDARs and ephrin receptors).52, This effect appears to be more rapid than the internalization of receptors, since the first changes to the composition at the glutamatergic synapse can be seen as early as 2 hours after Ab application. The effects of NMDAR-Abs also acutely interfere with the synapse’s ability to change in response to long-term potentiation protocols, limiting the ability of neuronal networks to remodel in response to activity.53
NMDAR-IgG Abs are mainly of the IgG1 subclass and they were shown to bind and activate the complement system in vitro, both in HEK cells expressing NMDARs and in neuronal cultures from the hippocampus.10 However, a role for NMDAR-Abs in the activation of complement has not been demonstrated in the limited numbers of biopsies or post mortem reports.53–55
Effects of NMDAR-Abs on more complex neuronal networks and CNS physiology have also been demonstrated. Intra-hippocampal infusions of patient IgG showed that NMDAR-Abs increase extracellular concentrations of glutamate and alter glutamatergic pathways and metabolism.56 Not all of the symptoms in NMDAR-Ab encephalitis are restricted to hippocampal functions. Indeed, NMDAR-Ab effects on neocortical networks were shown by an increase in corticomotor excitability after infusion of patient IgG into the prefrontal cortex.57
However, in vivo features which reproduce clinical features seen in affected patients are awaited. Indeed, many of the above studies used models that reduce the complexity of the hippocampal and neuronal networks and model the mechanism in vitro. Post mortem and biopsy studies of patients may offer more insights into the human pathophysiology or, alternatively, may only capture an end-stage disease. A detailed analysis of the cortical biopsy and autopsy pathology seen in NMDAR, VGKC, and GAD LE was recently published.53 The specimens showed infiltration with cytotoxic T-cells and lymphocytes, however this was much less commonly seen in cortex of NMDAR-Ab encephalitis patients.
Previous post mortem studies also showed prominent microgliosis in brain tissue from NMDAR-Ab positive patients.54 VGKC-complex and GADantibody encephalitis both showed a marked degree of neuronal loss within the hippocampus and evidence of axonal injury. These findings were not, however, observed in the tissue obtained from patients with NMDAR-Ab encephalitis patients. This would be in line with a mechanism by which antibodies primarily affect synaptic processing and act to modulate neuronal network activity in NMDAR-Ab encephalitis. Evidence of activation of the complement system was only present in VGKCcomplex Ab positive cases, highlighting this pathway as a possible explanation for loss of neurons within the hippocampus.
There is ongoing doubt whether Abs against intracellular targets can be truly pathogenic.4 However the MRI findings, pathology analyses and a recent report showing a decrease in cortical GABA concentrations in patients with GAD-Abs58 indicate the presence of a functionally significant inflammatory process. Whether the GAD-Abs are causing the processes or a simply a marker that coincides with unknown other Abs is currently difficult to assess.
More Questions to Answer
In 1957, Witebsky proposed criteria to determine if a disease is autoimmune in nature, based on Koch’s postulates describing the relationship between pathogens and disease. The criteria required an autoantibody-mediated autoimmune response, recognition of the target antigen, an autoimmune response which was inducible in animal models and animals immunized against the identified antigen developing a similar disease.
As reviewed above, few of these aspects have been shown for AIE to date. A recent case study by Hansen and colleagues however shows that intrathecal synthesis of NMDAR-Abs does not necessarily correlate with symptoms in patients.59 More research is needed to explain how Abs can be pathogenic, access the CNS, and how subsequently may lead to changes in the neuronal networks and cause the typical pattern of disease in patients. In particular, the recently described FBDS with its window of treatment opportunity might give further insight into the mechanisms involved in the disease progression. It will be very interesting to understand the localization of these early events and whether their treatment can halt the frequently observed progression to the amnesia and confusion which characterize AIE.24–26
It is rather difficult to model complex AID without an active immunization model, since each passive transfer relies on the fact that the animals’ immune system would respond to the human antibodies in the same way as the patients’. However, this is not always the case: passive transfer animal models of neuromyelitis optica (NMO) require the presence of human complement to fully show the pathogenic potential of NMO Abs.60
We are only just beginning to understand the complex mechanisms of disease caused by Abs within the CNS. Further research into early phases of disease and the Ab-mediated pathology might lead to novel targeted approaches which could complement the existing array of immunotherapy. In addition, formal prospective studies are needed to determine ideal treatment regimens for this potentially reversible cause of encephalitis.