Neuromyelitis optica (NMO, or Devic’s disease) is an autoimmune inflammatory disease of the central nervous system (CNS) that is typically associated with severe attacks of optic neuritis and myelitis and characteristically spares the brain early in the disease course. Although historically believed to be distinct from multiple sclerosis (MS) in that it presented with simultaneous or rapidly sequential optic neuritis and myelitis with a short interval, it is now believed that it usually begins with unilateral optic neuritis or myelitis and follows a relapsing course.1
Neuromyelitis optica (NMO, or Devic’s disease) is an autoimmune inflammatory disease of the central nervous system (CNS) that is typically associated with severe attacks of optic neuritis and myelitis and characteristically spares the brain early in the disease course. Although historically believed to be distinct from multiple sclerosis (MS) in that it presented with simultaneous or rapidly sequential optic neuritis and myelitis with a short interval, it is now believed that it usually begins with unilateral optic neuritis or myelitis and follows a relapsing course.1
In the past, the relapsing form of NMO was typically diagnosed as MS, based on the principle that relapsing CNS inflammatory disease was by definition MS. Over the last decade, however, studies of the clinical aspects, natural history, neuroimaging, pathology and immunology of NMO have established that it is distinct from prototypic MS.1,2 A specific immunoglobulin G (IgG1) autoantibody, NMO-IgG, was discovered in NMO patients3 that subsequently was shown to target the astrocytic water channel aquaporin-4 (AQP4).4 This advance has facilitated early recognition of NMO5,6 and its differentiation from MS, appreciation of a broader spectrum of manifestation, including limited forms of NMO (e.g. recurrent optic neuritis and recurrent transverse myelitis), and, perhaps most importantly, has facilitated breakthrough research in understanding NMO pathogenesis.
Diagnosis
Demographics and Clinical Characteristics
Although considered a rare disorder, NMO is almost certainly under-recognized and is frequently misdiagnosed as MS, idiopathic transverse myelitis, recurrent optic neuritis, or a connective tissue disease associated with myelitis (e.g. lupus myelitis). NMO usually manifests in adulthood, predominately in late middle age, but has been reported in children as young as 23 months of age7 and in adults as old as 80 years of age.8 There are two forms: monophasic and relapsing. Monophasic NMO presents with bilateral optic neuritis and myelitis within a short interval and no subsequent relapse of the index symptoms. Relapsing NMO, which is now accepted to be the most common form, accounts for approximately 80% of cases. The relapsing form of NMO is eight times more frequent in women than in men, and non-Caucasian individuals (African, Asian, and Hispanics) account for a larger proportion of NMO patients compared with MS, which has a predilection for individuals of European ancestry.9–12
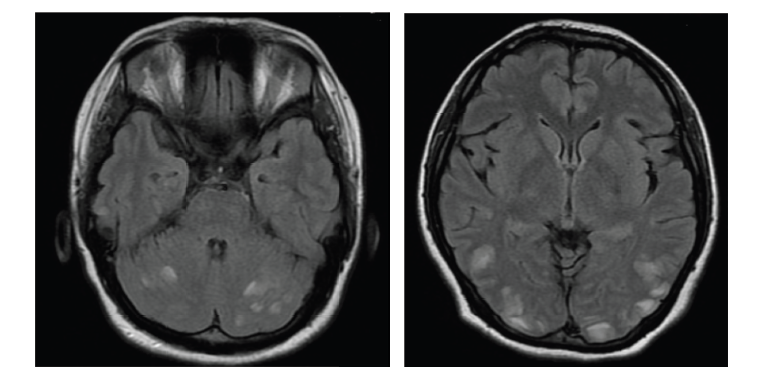
The correct diagnosis of NMO has been facilitated by formal diagnostic criteria that now incorporate NMO-IgG serological testing, which has become available worldwide and is offered by several specialty laboratories in academic medical centers. Wingerchuk et al. proposed that diagnostic criteria should not include the arbitrary interval between index episodes and the requirement of monophasic course, which were historically important distinguishing features from MS. In addition to optic neuritis and transverse myelitis that alone do not distinguish between NMO and MS, however, added specificity criteria were incorporated, the most important of which was the presence of a ‘longitudinally extensive spinal cord lesion.’ This is defined as a T2 signal lesion extending over three or more vertebral segments in the context of an acute myelitis attack on magnetic resonance imaging (MRI) scan (see Figure 1).1 Asymptomatic (but not symptomatic) brain lesions detected on MRI were allowed. NMO-IgG seropositivity3 and the observation that symptomatic brain lesions are compatible with a diagnosis of NMO led to the revision of the criteria in 200613 (see Table 1). The essential clinical features of NMO are optic neuritis and myelitis, which are usually more disabling in NMO than when they occur in the context of MS.1 Myelitis attacks are frequently ‘complete’ (symmetrical and with motor, sensory and sphincter involvement) rather than ‘partial.’ NMO optic neuritis attacks are more frequently bilateral with persisting visual deficits more severe than in MS. Brainstem syndromes are now recognized to be especially common. Severe and intractable hiccups, nausea, and vomiting lasting weeks to months may occur and may be the presenting symptom in patients with NMO.1,14,15 Occasionally patients may experience encephalopathy due to transient vasogenic brain edema and may be diagnosed as having posterior reversible leukoencephalopathy.16 Neurological disability in NMO is related to the attacks and a secondary progressive course is very uncommon, unlike MS.17
Neuroimaging
Spinal cord MRI in the acute phase of attack frequently demonstrates a central lesion, extending over three or more continuous vertebral segments. Gadolinium enhancement is usually present, but may be patchy. Acute lesions are usually hypointense on T1 imaging. Central cavities or longitudinally extensive cord atrophy may be present in later stages of lesion evolution.1 Confluent small chronic spinal cord lesions in MS patients may conglomerate and appear as a long lesion and may be mistaken for NMO; in NMO a spinal cord lesion usually results in a well-delimited long lesion when imaging is obtained acutely in the context of a disease attack (see Figure 1). Enhancing lesions of the optic nerve or optic chiasm may be observed in the acute phase of an optic neuritis attack (see Figure 2). Brain MRI lesions are detected in approximately 60% of NMO patients. Even though most brain lesions encountered in patients with NMO are non-specific and asymptomatic, lesions in the brainstem and hypothalamus appear to be relatively characteristic. Brain lesions occur more frequently in regions known to have high expression of aquaporin-4 (AQP4).18,19
Serological Testing
NMO-IgG is the disease biomarker found the in serum of NMO patients. In 2004, Mayo Clinic investigators identified a specific immunofluorescence pattern in a few NMO patients studied using a protocol to detect paraneoplastic syndrome autoantibodies. That same pattern had been recognized in other patients whose sera had been referred for paraneoplastic serology testing. When medical information on those patients whose sera had been identified strictly on serological basis was reviewed, the common clinical substrate was a history of severe and recurrent optic neuritis and/or myelitis. A prospective study was then conducted with clinically defined NMO patients; individuals with high-risk syndromes (recurrent longitudinally extensive transverse myelitis and recurrent optic neuritis); and with controls (MS and other neurological or autoimmune disease patients). The sensitivity and specificity of the test were 73 and 91%, respectively, for NMO versus MS, the entity with which NMO is most commonly confused.3 Several independent investigators have confirmed the presence of NMO-IgG using a variety of assays for anti-AQP4 antibody detection.20–24
NMO-IgG seropositivity and titer may have prognostic significance. In a prospective follow-up of patients with isolated transverse myelitis and in patients with recurrent optic neuritis but no transverse myelitis, seropositivity for NMO-IgG strongly predicted the occurrence of myelitis or optic neuritis relapse rate and worse disability. Patients with isolated or recurrent optic neuritis or isolated myelitis who are seropositive for NMO-IgG are now considered to be have an initial syndrome of NMO and should be treated accordingly.5,6 Serum levels of NMO-IgG may correlate with the disease evolution and treatment response. In a study with 96 samples from a series of eight NMO-IgG-positive patients (median follow-up 62 months) increasing NMO-IgG serum levels appeared to be associated with relapse (versus remission status) and with the CD19 cell counts in patients being treated with rituximab.25 In a different study, antibody titers were higher in patients with complete blindness in comparison with those with optic neuritis who experienced some degree of recovery and in patients with longer compared with shorter spinal cord lesions.26
Treatment
No clinical trial has been conducted primarily addressing treatment of NMO patients. The recommendations for both treatment of acute exacerbations and relapse prevention have been based on clinical trials that included patients with a variety of demyelinating diseases or on small open-label NMO small-series studies. Following an attack of NMO, high-dose intravenous methylprednisolone should be started as soon as possible. Based on a randomized controlled trial that included patients with NMO and acute transverse myelitis, and other non-randomized experience, plasmapheresis is recommended to treat attacks that do not respond to intravenous steroids.27–29
For long-term relapse prevention, immunosuppressive drugs are recommended rather than the immunomodulatory agents used for MS patients. Azathioprine30 (2.5mg/kg/day) with oral corticosteroids31 (for example, prednisone beginning at 60mg/day or every other day for nine months, subsequently decreasing to the lowest possible maintenance dose or discontinuation) and mycophenolate mofetil (2g/day) with oral corticosteroids are recommended initial therapies. For refractory cases, rituximab32,33 (1g intravenously twice separated by two weeks; repeated every six to nine months) and mitoxantrone34 (12mg/m2 every three months, with a maximum cumulative dose limited to 140mg/m2 primarily due to cardiotoxicity) have some evidence of efficacy in nonrandomized small series of NMO patients. Interferon (IFN)-beta is probably ineffective in NMO treatment.35,36
Future Prospects
The study of NMO has rapidly progressed due to the successful translation of the discovery of a specific biomarker into clinical practice and diagnostic criteria. The basic discoveries of the putative antigenic target of the immune response are achieving and will continue to achieve improved understanding of the physiopathology of NMO and development of new treatment. While it has not been absolutely established that NMO-IgG itself is pathogenic and not just a biomarker, there is strong circumstantial evidence that points to its pathogenic potential. One line of evidence is the concordance of microscopic pathology in human NMO with sites of high expression of AQP4 in the brain, particularly the perivascular foot processes of astrocytes and more specifically in the hypothalamus and periependymal regions of the brainstem and corpus callosum. A second line of evidence is the biological effect of NMO-IgG. An in vitro study demonstrated that NMO-IgG binds to the extracellular domain of AQP4 and leads to AQP4 endocytosis/degradation and complement-mediated cytotoxicity.37 NMO-IgG binding to astrocytes alters AQP4’s polarized expression and increases the permeability of the human blood–brain barrier, and can result in natural killer cell degranulation and astrocyte killing by antibody-dependent cellular cytotoxicity.38 Moreover, cells expressing AQP4 that were submitted to NMO-IgG in the absence of complement, have concomitant loss of sodium ion-dependent glutamate transport and loss of the excitatory amino acid transporter 2 (EAAT2), suggesting that EAAT2 and AQP4 coexist in the astrocytic membranes as a molecular complex.39 These observations on the downstream effects of NMO-IgG binding to AQP4 may suggest novel and important targets for effective intervention in this disease. ■