Neuromyelitis optica spectrum disorders (NMOSD) are a group of relapsing autoimmune diseases of the central nervous system. The clinical hallmarks of NMOSD are myelitis and optic neuritis; however, a wider clinical spectrum has been recognized.1 The majority of patients with NMOSD exhibit pathogenic immunoglobulin G autoantibodies against the astrocytic water channel aquaporin-4 (AQP4).2 While the understanding of seronegative NMOSD remains incomplete, AQP4-IgG-seropositive NMOSD (AQP4+ NMOSD) constitutes a well-defined disease entity and has emerged as a prototypic autoantibody-mediated autoimmune disorder.
The management of this disabling disease has long been unsatisfactory. However, following the recognition of AQP4-IgG, the B-cell-depleting anti-CD20 antibody rituximab has been successfully applied to NMOSD and remains one of the most frequently used treatment options to date.3,4 While rituximab is off-label for NMOSD in the USA and EU, in the last few years, four class I evidence-based drugs have been approved for attack prevention in AQP4+ NMOSD by the United States Food and Drug Administration (FDA) and European Medicines Agency (EMA), including the novel anti-CD19 B-cell-depleting monoclonal antibody inebilizumab.
In this article, we review the evidence for the use of inebilizumab in NMOSD and discuss differences between CD20- and CD19-mediated B-cell depletion, as well as the role of inebilizumab in the current treatment landscape of NMOSD. Due to the heterogeneity and incomplete understanding of seronegative NMOSD, this review article will focus on AQP4+ NMOSD.
Clinical presentation of neuromyelitis optica spectrum disorder
Patients with NMOSD most often present with myelitis, followed by optic neuritis.5–7 In contrast to the important differential diagnosis, multiple sclerosis (MS), both myelitis and optic neuritis tend to be longitudinally extensive in NMOSD (Figure 1).6,8–10 Myelitis is most often centromedullary (Figure 1b).11 Optic neuritis frequently occurs bilaterally.12 An important differential diagnosis for both bilateral optic neuritis and longitudinally extensive transverse myelitis is myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Formerly considered a variant of NMOSD, MOGAD is now recognized as a pathogenically distinct disease, and international diagnostic criteria have recently been proposed.13 A less frequent but relatively specific presentation of NMOSD is the area postrema syndrome, characterized by intractable hiccups, nausea and emesis.14 Furthermore, rarer presentations include other brainstem syndromes and cerebral syndromes with specific magnetic resonance imaging (MRI) features.1
Figure 1: Optic neuritis (a) and myelitis (b) in neuromyelitis optica spectrum disorder. Gadolinium-enhanced axial T1 (a) or sagittal T2 (b) magnetic resonance images
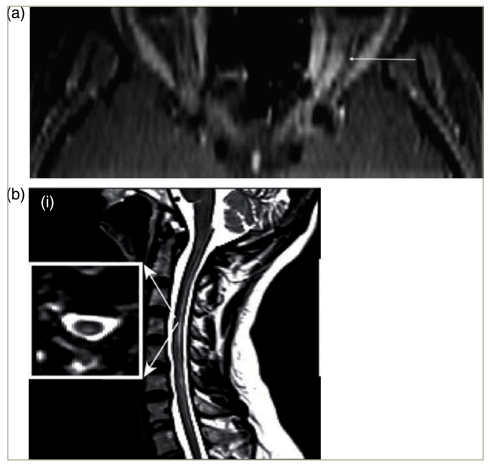
(a) Permission has been obtained from Klin Monbl Augenheilkd, Thieme Group for reuse of this figure. Citation: Mewes et al.8
(b) Reproduced with permission and without amendment from Chien et al.9
NMOSD can occur at any age, with a median onset age of around 35–40 years. Being a rare disease, its incidence differs according to geographic location and ethnicity. Women are strikingly more often affected by AQP4+ NMOSD than men (around 8:1).15,16
The natural disease course of NMOSD is characterized by recurring attacks. Remission after attacks is usually incomplete, and each single attack has the potential for severe persistent disability or even death.17 Importantly, a routine MRI is of limited value for monitoring attack risk in clinically stable patients due to the rarity of silent lesions, and alternative biomarkers are not yet clinically applicable.18 Even after long episodes of clinical stability and/or after long-lasting immunotherapy, AQP4+ NMOSD retains the potential to relapse.19–21 Therefore, attack-preventing therapy is paramount in NMOSD and, according to the currently available data, should be continued lifelong.22
Pathogenesis of neuromyelitis optica spectrum disorder
AQP4+ NMOSD constitutes a prototypic antibody-mediated autoinflammatory disease. While the aetiology (i.e. the reason why the disease occurs in certain individuals) of NMOSD remains obscure, it has well been demonstrated to be mediated by pathogenic anti-AQP4 IgG1 autoantibodies (Figure 2).2,23–28 The pathogenetic role of AQP4-IgG is supported by their high disease specificity, complement-activating potential and ability to induce NMOSD pathology in passive-transfer experiments.23 AQP4 is a water channel protein, most abundantly expressed on perivascular astrocyte end-feet. AQP4-IgGs are produced in the peripheral immune compartment.29 They traverse the blood–brain barrier at predilection sites and in states of increased permeability, such as in acute NMOSD attacks.30 Binding their antigen, AQP4-IgGs induce complement activation and lymphocyte as well as granulocyte and monocyte infiltration, leading to necrotic lesions hallmarked by astrocyte loss.31 Disability in NMOSD is acquired through acute attacks.17 Yet, evidence from biomarker studies implies potential attack-independent subclinical disease activity.32,33
Figure 2: Pathophysiology of aquaporin-4-immunoglobulin G-seropositive neuromyelitis optica spectrum disorder
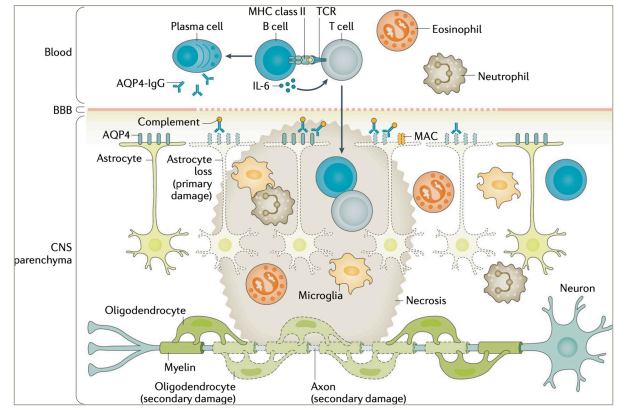
AQP4 = aquaporin-4; BBB = blood-brain-barrier; CNS = central nervous system; IgG = immunoglobulin G; IL = interleukin; MAC = membrane attack complex; MHC = major histocompatibility complex; TCR = T cell receptor.
Figure re-used with permission from Jarius S et al. Neuromyelitis optica. Nat Rev Dis Primers. 2020;22;6(1):85. DOI: 10.1038/s41572-020-0214-9.23
The role of B cells in neuromyelitis optica spectrum disorder
Several lines of evidence support a prominent involvement of B cells in the pathogenesis of AQP4+ NMOSD. B cells are lymphoid cells, whose development and maturation in the bone marrow and secondary lymphoid organs are hallmarked by immunoglobulin (Ig) gene segment rearrangement. B cells mediate highly specific immune responses against cognate, conformational antigens and maintain long-term adaptive immune memory. Their functions include antigen presentation, targeted T-cell response enabling, cytokine production and antibody production.34 Interleukin-6 (IL-6) is a pro-inflammatory cytokine, both produced (among other cell types) by B cells and inducing antibody production, that has been shown to promote disease activity in NMOSD.35 IL-6 blockade by satralizumab has been proven effective in AQP4+ NMOSD.36,37 B-cell depletion in NMOSD may exert its effects by reduction of both IL-6 levels and T helper 17 response.38 Antibodies are mostly produced by plasmablasts and plasma cells, developed from mature B cells. Average AQP4-IgG serum levels are higher during attacks than remission in NMOSD, and antibody removal by plasma exchange or immunoadsorption is an effective acute treatment in AQP4+ NMOSD.39,40 Peripheral blood plasmablast concentration has been shown to correlate with AQP4-IgG levels in AQP4+ NMOSD.35
Individual stages of B-cell development are characterized by specific patterns of cell surface antigen expression. Immature and mature B cells, as well as pre-B cells and memory B cells, express both the CD20 and CD19 surface antigens. In contrast, both early B-cell stages (pro-B cells) and plasmablasts and a subset of plasma cells express CD19 but not CD20.41 Therefore, anti-CD19 therapy bears the potential of higher effectiveness in AQP4+ NMOSD compared with anti-CD20 treatment.
Inebilizumab: pharmacology
Inebilizumab (Uplizna®, Horizon Therapeutics, Dublin, Ireland) is a monoclonal, humanized IgG1 antibody binding CD19. Inebilizumab is modified by afucosylation to increase Fc-gamma (Fcγ) receptor-binding affinity and antibody-mediated cytotoxicity.42 The median half-life of inebilizumab is 18 days, and reduced peripheral blood B-cell counts were maintained after 6 months in 94% of patients.43 Due to proteolytic degradation by ubiquitous enzymes, no dose adjustment is recommended for patients with impaired renal or hepatic function, although no studies have systematically addressed this population. Inebilizumab is administered intravenously in a dose of 300 mg at days 1 and 15, and afterwards every 6 months.44
Inebilizumab: preclinical data
Anti-CD19 monoclonal antibodies were first assessed in vivo in transgenic mice for human CD19, demonstrating rapid B-cell depletion in the bone marrow, spleen, lymph nodes and blood.45 The advantage of the mouse model is that it allows the assessment of B cells in tissue, while in humans, the effectiveness of B-cell depletion can only be assessed in peripheral blood, harbouring only a minority of an individual’s total lymphocytes. Anti-CD19-mediated B-cell depletion was shown to be mainly mediated by Fcγ receptor-dependent cytotoxicity (similar to anti-CD20 depletion), providing the rationale for glycoengineering inebilizumab to enhance this interaction.45 Furthermore, unlike in common anti-CD20 models, anti-CD19 B-cell depletion induced a strong decrease in Ig levels, probably reflecting partial plasma cell depletion.45 In theory, this might convey both higher effectiveness and infection risk.
Inebilizumab was shown to induce profound B-cell depletion in the blood, spleen and bone marrow in CD19 transgenic mice.42,46 This B-cell depletion was not complement-dependent, which might imply the maintained effectiveness during complement inhibition.42 In a mouse model of lupus-like autoimmunity, inebilizumab depleted autoantibody-producing cells in the spleen and strongly reduced serum autoantibody levels.46 In contrast, autoantibody-producing cells were not depleted in the bone marrow, and the total serum Ig levels were almost unaltered.46
Inebilizumab: clinical trial data in neuromyelitis optica spectrum disorder
The N-MOmentum trial (A Clinical Research Study of Inebilizumab in Neuromyelitis Optica Spectrum Disorders. ClinicalTrials.gov identifier: NCT02200770) was a double-blind, randomized phase II/III trial comparing inebilizumab with a placebo in adult patients with NMOSD.47 176 patients received inebilizumab, and 56 patients were included in the placebo group. Overall, 92% of all study participants were AQP4-IgG-seropositive. The primary endpoint was the time to an adjudicated NMOSD attack within 197 days (randomized controlled period), which was significantly shorter in the inebilizumab group.47 The randomized controlled study period was terminated early due to clear efficacy. The hazard ratio for occurrence of an attack was 0.27, and the number needed to treat was 3.7. Inebilizumab was approved for attack prevention in adult patients with AQP4+ NMOSD by the FDA and EMA in 2020 and 2022, respectively.
Secondary endpoints were worsening of the Expanded Disability Status Scale (EDSS) score, binocular low-contrast visual acuity, number of new active MRI lesions and NMOSD-related hospitalizations. In patients treated with inebilizumab, NMOSD worsening and NMOSD-related hospitalizations, as well as new active MRI lesions, occurred less often.47 Binocular visual acuity change was not different between inebilizumab and placebo (secondary endpoint), yet patients treated with inebilizumab were less likely to experience optic neuritis.47
Disability, as measured by the EDSS, improved in inebilizumab-treated, but not in placebo-treated, patients in the N-MOmentum trial, and the number of patients with a favourable modified Rankin scale (<2) score at the end of the trial was higher in the inebilizumab group.48 Disability scores remained stable during the open-label extension over a total of >4 years.49
Biomarkers of disease activity were assessed in the N-MOmentum trial, including the number of active MRI lesions and serum glial fibrillary acidic protein (sGFAP). The cumulative number of active (i.e. gadolinium-enhancing) MRI lesions in the brain, optic nerve or spinal cord after baseline was lower in the inebilizumab group (mean 2.3 versus. 1.7). As subclinical MRI activity is rare in NMOSD, this might be interpreted as a paraclinical surrogate of the reduced incidence of attacks.18,50 GFAP is an astrocytic protein and a promising, biologically plausible candidate biomarker for disease activity in AQP4+ NMOSD.33 GFAP levels increased during the attack in the placebo group but not in most inebilizumab-treated patients.51 Absolute sGFAP levels decreased, compared with baseline, only in the inebilizumab but not in the placebo group.51 Furthermore, high levels of serum neurofilament light chain, a marker for neuroaxonal injury, occurred less frequently in inebilizumab-treated, compared with placebo-treated, patients with NMOSD.52
Sensitivity and subgroup analyses of the N-MOmentum trial revealed no dependency of the attack risk reduction in the inebilizumab group on the type of attack, baseline disability, ethnicity, treatment history and prior disease course.53 In a detailed subgroup analysis of the Asian participants in the N-MOmentum trial, no relevant differences were detected regarding the efficacy and safety profile compared with the whole group.54 Of note, the predefined attack definition in N-MOmentum was rigorous and allowed the inclusion of MRI findings in clinically inconclusive cases.55 Consequently, the reduction of the attack risk by inebilizumab is similar, regardless of whether adjudicated attacks (confirmed by the adjudication committe) or investigator-reported attacks (not confirmed by the adjudication committe) were analyzed.53,56
Subgroup analyses of the AQP4-IgG-seronegative participants in the N-MOmentum trial were limited due to the presence of only four patients in the placebo group (13 with inebilizumab; total of three attacks and all in inebilizumab-treated patients). Numerically, the hazard ratio for time to attack onset in inebilizumab- versus placebo-treated patients was lower in the AQP4+ compared with the total group.47 No valid comparison of AQP4-IgG-seropositive and AQP4-IgG-seronegative patients was possible regarding the primary endpoint. Seven AQP4-IgG- N-MOmentum participants were myelin oligodendrocyte glycoprotein (MOG)-IgG seropositive (one placebo participant and six inebilizumab participants). 57 When considering the open-label extension, there was a reduction in annualized attack rates after initiating inebilizumab in AQP4-IgG- and MOG-IgG+ patients, as well as in double-seronegative patients, compared to before its initiation.
The frequency of adverse events and serious adverse events in the N-MOmentum trial was similar in the inebilizumab group and placebo group.47 Most frequent adverse effects included urinary tract infections, infusion-related infections, arthralgia, headache and nasopharyngitis. No deaths occurred in the randomized controlled period, while two deaths occurred in the open-label extension (respiratory insufficiency after an NMOSD attack and aetiologically undetermined encephalitis, respectively). Adverse events numerically more frequent in the inebilizumab group included, among others, urinary tract infections and headache.47 Results regarding adverse events from phase I studies in patients with MS and systemic sclerosis were largely in line with these findings.58,59
It is well established that anti-CD20-treated patients display a weakened humoral vaccine response, including against anti-severe acute respiratory syndrome coronavirus type 2 (SARS-COV2) vaccines, although a cellular response is preserved.60,61 Sufficiently large examinations of patients receiving inebilizumab have not been published to date. In one small study, anti-spike protein antibodies were detected in one inebilizumab-treated patient after SARS-COV2 infection but in none of the three after anti-SARS-COV2 vaccination.62
The reduced attack risk in inebilizumab-treated patients with AQP4-IgG+ NMOSD was present for at least 4 years in data from the randomized controlled period as well as from the open-label extension of the N-MOmentum trial.49 Most attacks took place in the first year of treatment, suggesting that the efficacy of inebilizumab might possibly increase with a longer treatment duration.49
Inebilizumab: monitoring and safety
Prior to the initiation of treatment with inebilizumab, chronic or latent infections (such as hepatitis B infection, hepatitis C infection, human immunodeficiency virus [HIV] infection or tuberculosis) should be excluded. Inebilizumab is contraindicated in patients with active hepatitis B infection or active or untreated latent tuberculosis due to the risk of reactivation. Due to both the increased risk of infections and the limited efficacy of vaccinations under B-cell-depleting treatment, immunizations according to applicable guidelines should be completed before the initiation of inebilizumab. Live vaccinations should not be performed during B-cell depletion.
Next to screening for infections, a differential blood cell count, peripheral blood B-cell count and serum Ig levels should be obtained before starting inebilizumab. In anti-CD20-treated individuals, low IgG levels before treatment are predictive of hypogammaglobulinaemia.63 During inebilizumab treatment, serum levels of all Ig classes decrease.64
As low Ig levels and lymphopaenia might develop under therapy and convey an increased risk of infections, they should be tested before each inebilizumab dose.47,63 While no case of definite progressive multifocal leukoencephalopathy has so far been described in any inebilizumab-treated patient, anti-CD20-treated patients with severe lymphopaenia are considered at an increased risk, and it is plausible to assume this also for anti-CD19.65
Prior to each inebilizumab dose, patients should be screened for active infection and receive a premedication of methylprednisolone, diphenhydramine and acetaminophen/paracetamol to avoid infusion reactions. Patients should be monitored on-site during and until at least 1 h after the infusion.44
Monitoring B-cell depletion in patients is generally incomplete, as only about 2% of the total lymphocyte pool is located in the blood and therefore accessible to routine analysis.66 Furthermore, B-cell counts are commonly determined using CD19-labelling in anti-CD20-treated patients to avoid interactions with the cell-bound biological. Conversely, it is reasonable to use CD20 to monitor B-cell depletion during the inebilizumab treatment. Though not mandatory, regular B-cell counts may be helpful in identifying non-/no-longer-responders, for example, with anti-drug antibodies (ADAs). ADAs were detected in 14.7% of patients in the inebilizumab trial.64 Moreover, the extent of B-cell depletion after 6 months of inebilizumab treatment is associated with clinical and imaging disease activity in NMOSD.64 Specifically, patients with 4/µL or less CD20-positive B lymphocytes had a lower annualized attack rate and less frequently had new or enlarging MRI lesions.64
There are currently no substantial data on the safety of inebilizumab during pregnancy and breastfeeding. As a class G antibody, inebilizumab is expected to transverse the placenta and to be excreted into breast milk, causing B-cell depletion in the foetus or newborn. Therefore, women of childbearing age should use effective contraception during and until at least 6 months after inebilizumab treatment.44 No studies have addressed the paediatric use of inebilizumab to date, and inebilizumab is only approved for the treatment of adult patients with AQP4+ NMOSD.44
CD19- versus CD20-mediated B-cell depletion in neuromyelitis optica spectrum disorder
No direct comparison between inebilizumab and rituximab in patients with NMOSD is available. Therefore, most considerations are theoretical or based on preclinical data. The efficacy of inebilizumab for attack prevention in AQP4+ NMOSD is supported by class I evidence, as discussed earlier. Several observational studies as well as one small (N=38) double-blind, randomized placebo-controlled trial and its open-label extension support the efficacy of rituximab to prevent NMOSD attacks in AQP4+ NMOSD.3,67–69 One open-label randomized trial suggests the superiority of rituximab over azathioprine.70 Observational evidence implies the possible efficacy of rituximab also in paediatric NMOSD.71 In the USA and EU, inebilizumab, but not rituximab, has been approved for maintenance treatment of adult AQP4+ NMOSD.
The depletion of B cells in early stages by inebilizumab, but not anti-CD20 antibodies,(Figure 3) might theoretically provide an additional mechanism of disease control, based on NMOSD pathogenesis, as dysregulation of early B-cell immunotolerance checkpoints has been demonstrated in NMOSD.73 More generally, anti-CD19 depletion of pre-B cells, prior to receptor selection, might deplete pathogenic B-cell precursor clones.41 Furthermore, CD19-mediated B-cell depletion may be more profound and longer lasting compared with CD20-mediated depletion in the mouse model, possibly due to the loss of B cells in earlier stages.42,45 However, (1) this was not demonstrated in humans and (2) might as well be interpreted as an advantage for CD20 therapies, regarding safety considerations.
Figure 3: Expression of CD19 and CD20 by B cells in different developmental stages
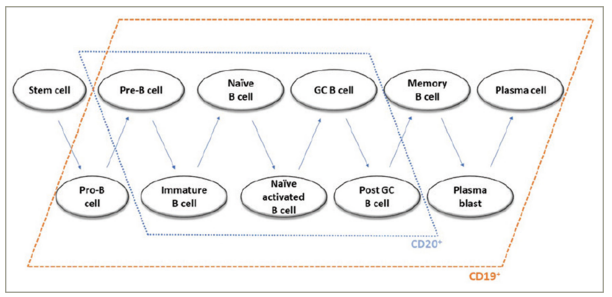
GC = germinal centre.
Reproduced with permission from Siebert et al.72 Copyright © 2021 Clarivate or its licensors. All rights reserved.
The depletion of plasmablasts and, partly, plasma cells by inebilizumab, but not rituximab, might induce a decrease in autoantibody titres, as suggested by animal studies.46 Notably, memory B cells, which can present antigens and may develop into AQP4-IgG-secreting plasma cells, carry the CD19 and CD20 surface molecules (Figure 3).74,75
A small proportion of T lymphocytes is CD20 positive, but CD19 negative, and efficiently depleted by rituximab.76 Whether this contributes to the anti-autoinflammatory effects of CD20 depletion, however, remains unclear.
B-cell depletion by both rituximab and inebilizumab partly depends on binding to Fc receptor III A (FCGR3A) on natural killer cells. While the efficacy of rituximab is reduced in carriers of a polymorphism in the FCGR3A gene (F158), the efficacy of inebilizumab is not relevantly affected by this polymorphism.77 However, in another analysis, 10/10 inebilizumab-treated patients who did not reach low B-cell counts during the first 6 months of treatment had the F/F genotype.64
Compared with the chimeric antibody rituximab, the humanized antibody inebilizumab is less likely to cause infusion-related reactions, and a inebilizumab dose can be infused over a shorter time (~1.5 h) than a rituximab dose (~4 h for 1 g).47,68
Observational clinical evidence is derived from patients with NMOSD switching from rituximab to inebilizumab. Seventeen patients, 16 of whom are AQP4-IgG+, receiving inebilizumab in the N-MOmentum trial (randomized controlled or open-label period) had been previously treated with rituximab.78 Three of these patients experienced an attack while on inebilizumab. Notably, none of the seven patients with breakthrough attacks during rituximab treatment experienced an attack while on inebilizumab. However, patients with a history of rituximab treatment more frequently had infectious adverse events while treated with inebilizumab, including severe infections.78 Moderate (<500 mg/dL; 23%) and severe (<300 mg/dL; 12%) IgG deficiency occurred more frequently in inebilizumab-treated patients previously receiving rituximab.78
As long-term data on CD19-mediated B-cell depletion are limited, it cannot be excluded to date that after years of treatment the safety profile might diverge from CD20-mediated B-cell depletion. It is an important task for future cohort/registry studies to address this aspect.
The role of inebilizumab in the current treatment landscape of neuromyelitis optica spectrum disorder
Currently, four immunomodulatory drugs have been approved by the FDA and EMA for the treatment of AQP4-IgG+ NMOSD: the inhibitors of the terminal complement cascade, eculizumab and ravulizumab, and the IL-6-receptor inhibitor satralizumab and inebilizumab.36,37,47,79,80 The Japanese drug regulatory authority approved rituximab for attack prevention in NMOSD in 2022.68 No head-to-head comparisons between these drugs exist. Older, off-label options, such as azathioprine or mycophenolate mofetil, are considered less effective but continue to play a role in special situations and when the biologicals are not available due to economic reasons. It should also be noted that prices diverge largely between biologicals used for the treatment of NMOSD, including between rituximab and inebilizumab (the German list of price for MabThera [500 mg rituximab, F. Hoffmann-La Roche AG, Basel, Switzerland] is €2,298.82 [twice 1 g yearly = €9,195.28] and for Uplizna [300 mg inebilizumab, Horizon Therapeutics, Dublin, Ireland] is €55,913.21 [twice 300 mg yearly = €111,826.42]).81
Consensus statements that incorporate recently approved drugs count them as first-line options, without general preference for one drug or the mechanism of action.22,82 The choice for a specific medication has therefore depended on individual patient characteristics as well as the preference of the patient and treating physician. Satralizumab is also approved for patients aged <18 years. Eculizumab, but not ravulizumab, is only approved for relapsing disease (i.e. not after the first attack).
Many patients with NMOSD are treated with rituximab. Therefore, the question arises whether it is reasonable to switch these patients to inebilizumab. While research on this aspect is scarce to date, the following statements are backed by emerging consensus: (1) patients being stable on rituximab need not be switched; (2) if rituximab is discontinued, for example, due to reasons of insurance coverage, it might be effective to switch to inebilizumab, but the risk of infections should be carefully considered; (3) though available data imply that inebilizumab may also be effective in patients who experienced an attack while on rituximab treatment, this is based on few observations, and the availability of several alternative drugs generally favours a switch of the mechanism of action.22,78
Conclusions
The anti-CD19 monoclonal antibody inebilizumab is effective and on-label for attack prevention in adult patients with AQP4+ NMOSD. It is considered a first-line option, alongside IL-6-receptor blockers and inhibitors of the terminal complement cascade. While the anti-CD20 monoclonal antibody rituximab is off-label for NMOSD in most countries, there is no evidence for the superiority of inebilizumab over rituximab regarding the efficacy or safety. Infections are the most important adverse effects of B-cell depletion, and serum Ig levels and differential leucocyte counts need to be carefully considered. While long-term experience with inebilizumab is limited to date, current evidence suggests that maintenance therapy in AQP4+ NMOSD might generally be necessary for life. As anti-CD20-mediated B-cell depletion has emerged as an important treatment principle in a large number of autoimmune and neoplastic disorders, including MS, intriguing research questions arise regarding the precise immunological and clinical sequelae of anti-CD19 therapy.