
Why this topic matters
Autoimmune psychosis (AP) is conceptualized as a psychosis-dominant form of autoimmune encephalitis (AE). In contrast to ‘typical’ AE, in which seizures, impaired consciousness and focal deficits rapidly declare a neurological syndrome, patients with AP can initially present to psychiatric services with apparently isolated psychotic or mood symptoms. Overt neurological signs may be subtle or evolve only later. Failure to recognize this autoimmune aetiology risks prolonged exposure to ineffective antipsychotic treatment, potentially serious adverse effects and missed opportunities for disease-modifying immunotherapy. Increased awareness of AP among both neurologists and psychiatrists is therefore crucial to shorten diagnostic delay and improve outcomes.
What is autoimmune psychosis?
Acute encephalitis is classically defined as rapidly progressive brain inflammation causing altered mental status, usually developing over days to a few weeks. It affects about 5–10 people per 100,000 each year and is most often infectious.1 Over the past two decades, non-infectious, immune-mediated forms of encephalitis have been recognized, grouped together as AE.1
Within this spectrum, AP describes patients with subacute (≤3 months) onset or worsening of psychotic or severe mood symptoms in whom immune mechanisms are likely to play a central role.2 Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis is the best-known example, but AP is not limited to any single antigen. The key point is that psychiatric features dominate at presentation and classical encephalitis criteria may not yet be met.
Pollak et al. proposed a practical three-step diagnostic scheme.2 ‘Possible AP’ refers to a rapidly evolving psychosis or major mood episode accompanied by at least one feature suggesting organic disease, such as recent tumour, catatonia or dyskinesias, marked cognitive impairment, new-onset seizures, autonomic instability or an unusually severe reaction to antipsychotics. ‘Probable AP’ adds supportive findings on cerebrospinal fluid (CSF), magnetic resonance imaging (MRI) or electroencephalogram (EEG), for example, CSF pleocytosis, medial temporal lobe fluid attenuated inversion recovery (FLAIR) lesions, oligoclonal bands or encephalopathic EEG changes. ‘Definite AP’ requires detection of a relevant antineuronal antibody in CSF, with paired serum testing when available. These categories are helpful for structuring clinical reasoning but are consensus–based rather than prospectively validated, and they do not replace formal AE criteria.
From an immunological standpoint, AP usually reflects either cell-surface or synaptic antibody-mediated disease, as in NMDAR or leucine-rich, glioma-inactivated 1 (LGI1) encephalitis, which tends to respond well to immunotherapy. Furthermore, antibodies against gamma aminobutyric acid (GABA)A (GABAA) receptors, metabotropic glutamate receptor 5, immunoglobulin-like cell adhesion molecule 5 (IgLON5) and other targets have been linked to syndromes in which psychosis, behavioural change or severe sleep disturbance can dominate at onset or recur throughout the course.3–5 On the other hand, T-cell-mediated paraneoplastic syndromes associated with antibodies targeted intracellular antigens, such as anti-Hu antibodies or anti-Ma1/2 antibodies, in which psychiatric symptoms may be prominent but often coexist with other neurological deficits and outcomes, are typically poorer. Small studies in related antibody-positive autoimmune encephalitis suggest broader disturbances in circulating B- and T-cell subsets, but such observations cannot yet be assumed to apply to all patients labelled clinically as having AP, particularly when neuronal antibodies are not detected.6 These observations support the view that AP reflects a broader systemic derangement of adaptive immunity rather than a purely ‘brain-limited’ process.
Psychopathology and clinical course
From a descriptive psychopathology perspective, AP and other psychosis-predominant forms of AE typically show a pleomorphic and often rapidly fluctuating picture. Patients may present with combinations of persecutory or grandiose delusions, auditory and visual hallucinations, thought disorder, disorganized or bizarre behaviour and marked mood lability. Anxiety, panic attacks, irritability, aggression and severe insomnia are frequent. Short-term memory impairment, attentional deficits and slowed information processing are common and often striking given the brief duration of illness and the person’s previous level of functioning.
Catatonia is particularly important to recognize. It may manifest as mutism, posturing, stupor, echolalia or echopraxia, or as an excited form with agitation and stereotyped movements. Orofacial dyskinesias, choreoathetoid limb movements or a mixture of dystonia and myoclonus can evolve over days or weeks, sometimes after the earliest psychiatric symptoms. In a recent cohort of patients meeting consensus criteria for AP and treated with immunotherapy, hallucinations were recorded in more than four-fifths of cases, delusions in around three-quarters, disorganized speech in two-thirds, severe cognitive dysfunction in a similar proportion and catatonia in well over half, underlining how prominent and polymorphic the psychiatric syndrome can be.7
Although the majority of patients with antibody-positive AE eventually develop overt neurological signs such as seizures, dyskinesias, speech disturbance or reduced level of consciousness, these features may emerge only after days or weeks of apparently isolated psychiatric illness.2 A smaller subgroup maintains a predominantly psychotic or behavioural presentation throughout the course.
When should I suspect autoimmune psychosis?
In practice, clinicians need quick cues that move AP up the differential. Several clinical ‘red flags’ are particularly helpful in a first-episode or rapidly worsening psychosis.2 These include a preceding infectious-like prodrome; a new severe headache or marked change in a pre-existing headache pattern; rapid escalation of psychiatric symptoms over days to weeks; paradoxical or severe reactions to antipsychotics, including features of neuroleptic malignant syndrome; insufficient therapeutic response despite adequate antipsychotic trials; the development of movement disorders such as catatonia or characteristic dyskinesias; focal neurological signs; reduced level of consciousness; prominent autonomic dysfunction; language disturbances including aphasia, mutism or dysarthria; new-onset seizures; evidence of a current or recent tumour; otherwise unexplained hyponatraemia; coexisting systemic autoimmune disease (e.g. systemic lupus erythematosus or autoimmune thyroiditis) and sensory symptoms such as paraesthesias. The overall pattern, tempo and clustering of such features are often more informative than any single item in isolation.
How should I investigate?
Once AP or psychosis-dominant AE is on the radar, investigations should be organized rather than piecemeal. As with AE more generally, the diagnosis is ultimately one of exclusion: structural lesions, metabolic and toxic encephalopathies, primary psychiatric disorders, substance misuse and infectious causes all need to be considered and reasonably ruled out. A practical starting point is basic laboratory testing, including full blood count, electrolytes with close attention to sodium, renal and liver function, inflammatory markers and targeted infectious serology or polymerase chain reaction (PCR) guided by the history and local epidemiology.
A structured, stepwise diagnostic work-up is essential once AP is suspected. Initial laboratory investigations should include routine blood tests such as electrolytes (with careful attention to serum sodium), full blood count and basic metabolic profile, alongside infectious screening guided by the clinical context (Figure 1).
Figure 1: Suggested diagnostic pathway for subacute psychosis with possible autoimmune aetiology
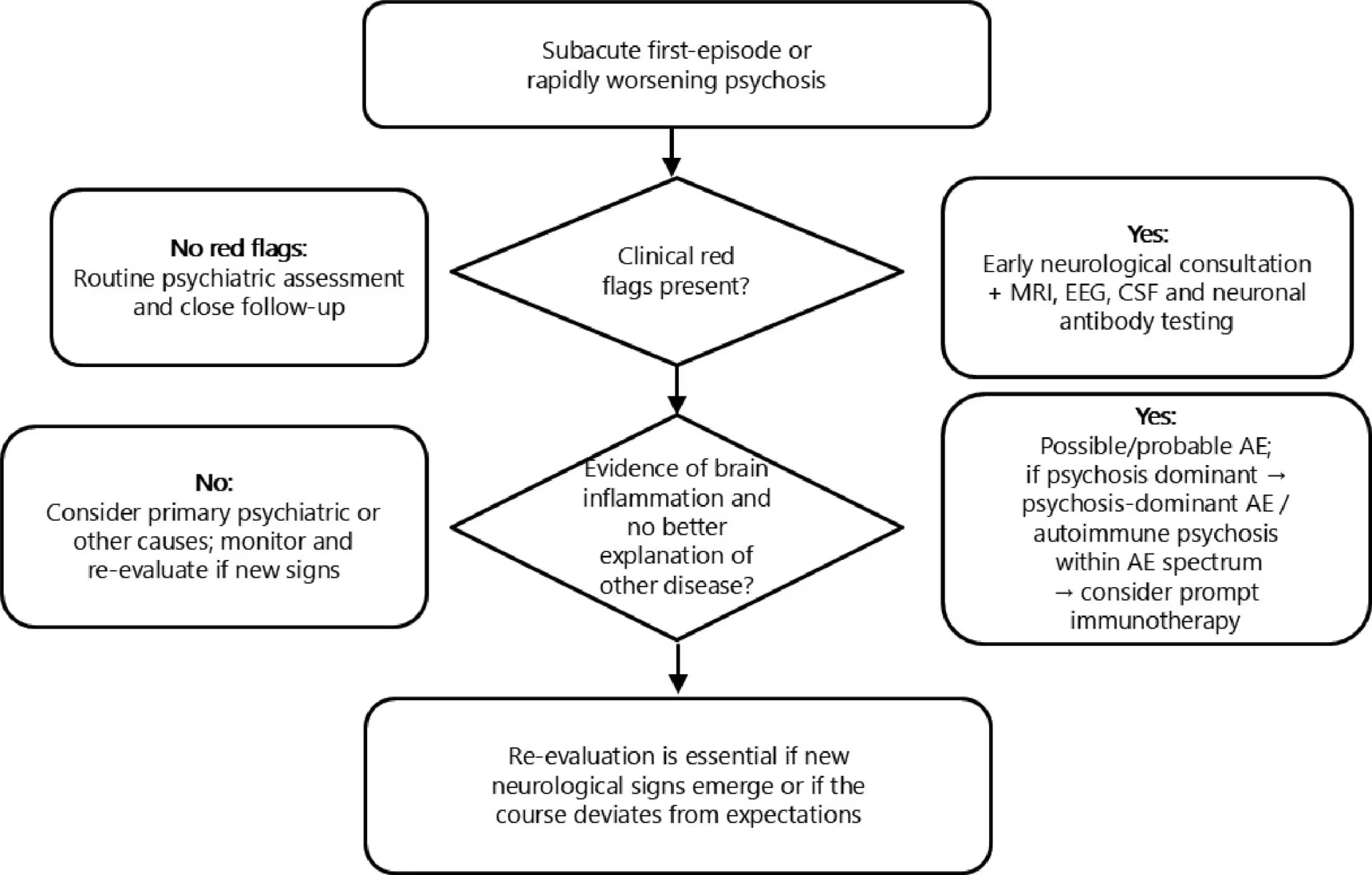
Patients presenting with first-episode or rapidly worsening psychosis are first screened for clinical red flags, including very rapid symptom evolution, catatonia or prominent dyskinesias, autonomic instability, seizures, focal neurological signs, new severe headache, hyponatraemia, other autoimmune disease or recent tumour. In the absence of red flags, routine psychiatric assessment and close follow-up are usually appropriate. When one or more red flags are present, early neurological consultation, brain MRI, EEG, CSF analysis and neuronal antibody testing are recommended. If this work-up demonstrates evidence of brain inflammation and no better explanation is found, the patient is considered to have possible or probable AE; when psychosis is the dominant feature, we describe this as psychosis-dominant AE or autoimmune psychosis within the AE spectrum and consider prompt immunotherapy. Re-evaluation is essential if new neurological signs emerge or if the course deviates from expectations
AE = autoimmune encephalitis; CSF = cerebrospinal fluid; EEG = electroencephalogram; MRI = magnetic resonance imaging.
Brain MRI with and without contrast is recommended, ideally early.1 Many patients with AP have normal MRI findings, but subtle cortical, mesial temporal or multifocal signal changes may be seen and can support the diagnosis. A normal scan should not divert attention away from an autoimmune process when clinical suspicion is high.
EEG is a low-risk, high-yield investigation. It can reveal non-convulsive seizures, diffuse slowing compatible with encephalopathy or patterns such as ‘extreme delta brush’ that are highly suggestive of NMDAR antibody encephalitis. In some cases, EEG abnormalities appear before changes on MRI.
CSF analysis should include cell count, protein and oligoclonal bands, as well as targeted infectious studies where indicated.1 CSF antineuronal antibody testing, preferably using a reliable cell-based assay, often carries higher specificity than serum testing for cell-surface antibody syndromes, whereas for some antibodies, such as LGI1, serum may be more sensitive.8 Isolated low-titre neuronal antibodies in serum, particularly when the clinical picture and investigations are otherwise unremarkable, should be interpreted with caution and ideally discussed with a specialist laboratory or autoimmune neurology service.9 Serum and CSF neurofilament light chain levels reflect axonal injury and are often higher in AE than in controls, with modest correlations with disease severity and functional outcome.10 At present, these assays are mainly available in specialist centres and should be interpreted as supportive evidence of neuroinflammation or neuroaxonal damage rather than as stand-alone diagnostic tests for AP.10 Elevated levels of kappa free light chains may also indicate intrathecal immunoglobulin synthesis in patients with psychiatric disorders, and thus help evaluate an autoimmune basis for psychiatric syndromes.11
Finally, appropriate tumour screening – for example, pelvic imaging in young women, testicular ultrasound in young men and chest or abdominal imaging in older patients – is important in view of paraneoplastic associations. Repeating screening may be justified when suspicion persists despite an initial negative work-up.
How should I treat?
Immunotherapy is the cornerstone of treatment once AP is considered likely and infectious causes have been excluded or are being appropriately treated.1 Clinicians should not wait for antibody results if the overall picture strongly supports an autoimmune aetiology. First-line regimens typically combine high-dose corticosteroids with intravenous immunoglobulin and/or plasma exchange, chosen according to local practice and patient-specific factors.1 In refractory or relapsing disease, escalation to second-line agents, such as rituximab and cyclophosphamide, can be considered, following protocols used for AE more broadly. 1Throughout treatment, careful monitoring is required for steroid-related psychiatric worsening, metabolic disturbance and heightened infection risk, with prompt adjustment of therapy when necessary.
Psychiatric symptoms still require active management. No antipsychotic has proven superiority in AP, but starting with low doses, titrating slowly and preferring atypical agents helps to reduce extrapyramidal side effects. For the longer term, steroid tapering over several months is often combined with oral steroid-sparing agents such as azathioprine or mycophenolate in patients at high risk of relapse. Clinicians should watch carefully for fever, rigidity, autonomic instability and creatine kinase elevation, given the higher risk of neuroleptic malignant-like reactions in AE. Benzodiazepines are often useful for anxiety, agitation and especially catatonia, and can help stabilize behaviour while immunotherapy takes effect. In severe, life-threatening or refractory catatonia, electroconvulsive therapy has shown benefit in many reported cases of NMDAR antibody encephalitis and should be considered where available.12
Supportive care should not be underestimated. Admission to a monitored or intensive care environment is appropriate when there is major dysautonomia, profound catatonia or severe agitation with medical instability. Attention to nutrition, hydration, prevention of aspiration, sleep–wake regulation and minimization of delirium triggers can all influence long-term outcome.
In the large cohort of patients with AP reported by Ramirez-Bermúdez et al., the majority of those who received immunotherapy showed marked reductions in hallucinations, delusions and catatonia alongside improved independence in daily activities.7 Similar response rates, typically in the range of roughly two-thirds to four-fifths, have been described in other autoantibody-associated psychiatric syndromes, particularly when treatment is initiated before the illness has become chronic or antipsychotic-refractory.7
Pitfalls and communication
Several recurring pitfalls are worth highlighting. First, existing diagnostic schemes for AP are designed to support clinical reasoning, not to replace it.2 Their sensitivity early in the course of disease is imperfect. Over-reliance on checklists can therefore leave atypical but genuine autoimmune cases unrecognized, especially in young patients with apparently ‘pure’ psychiatric presentations.
Second, antibody tests require cautious interpretation. Low-titre serum antibodies without supportive clinical, CSF or imaging evidence can occur as incidental findings.13 Overinterpreting such results risks mislabelling patients and exposing them to unnecessary immunosuppression.13 Conversely, failing to perform CSF analysis, MRI or EEG in patients with clear red flags can delay life-saving treatment. A balanced view that weighs pre-test probability against test performance is essential.
Third, communication with patients and families needs care. AP should not be presented either as a guaranteed route to full recovery or as a way to discard an unwanted psychiatric diagnosis. Autoimmune and primary psychiatric mechanisms may coexist, and diagnostic labels can evolve over time. Honest explanations of uncertainty, regular reassessment and shared decision-making about investigations and treatment help maintain trust during what is often a prolonged and emotionally demanding illness.
Take-home messages
AP offers a useful lens for thinking about patients who present with subacute psychosis but have features that do not fit a straightforward primary psychiatric disorder. Clinicians should maintain a low threshold for suspecting AP when there is a short prodrome, rapid deterioration, catatonia or dyskinesias, dysautonomia, unusual reactions to antipsychotics, seizures, hyponatraemia or evidence of systemic autoimmunity. CSF studies, EEG, MRI and antibody testing form the core diagnostic toolkit, with CSF antibodies generally providing the most specific evidence.1 Early, adequately dosed immunotherapy, combined with cautious use of psychotropic drugs and good supportive care, can lead to substantial recovery in many cases. Recognizing common pitfalls and communicating uncertainty openly are crucial to safe and effective practice.







